ハイライト
せん断応力誘導内皮細胞HEG1シグナル伝達は、血行動態と全身的な血圧制御を結ぶ新たな規制経路を代表します。高血圧患者では、血管内皮へのせん断応力の減少により、血漿中のHEG1レベルが著しく低下します。マウスでの内皮特異的なHeg1欠損は、血圧の上昇、内皮依存性血管拡張の障害、およびNO産生の乱れを引き起こします。メカニズムは、HEG1によるPHACTR1分解の制御に関与し、SP1依存性eNOS転写を制御します。
背景
高血圧は、世界中で心血管疾患、脳卒中、腎機能不全の主要な修正可能なリスク要因であり続けています。複数の降圧薬が利用可能であるにもかかわらず、多くの患者が適切な血圧管理を達成できないため、新たな治療標的の必要性が強調されています。内皮細胞は、全体の血管系を覆い、特に壁面せん断応力を含む血液力学的力の重要な機械センサーとして機能し、血管トーンと血圧恒常性を制御する生化学的信号を翻訳します。
血流せん断応力は、内皮表面に作用する摩擦力であり、そのメカニズムはまだ完全には理解されていませんが、血管拡張を刺激します。このメカノトランスダクション経路の障害は、高血圧と動脈硬化性心血管疾患の特徴である内皮機能不全に寄与します。せん断応力感知から一酸化窒素(NO)産生までの分子中間体を理解することは、血圧管理の実行可能な標的を明らかにする可能性があります。
心臓発生タンパク質EGF様ドメイン1(HEG1)は、以前に血行動態と心血管表現型と関連している内皮由来膜タンパク質です。疫学データは、HEG1発現と心血管リスク要因との逆相関を示唆していますが、この関連のメカニズム的基礎は定義されていません。本研究では、内皮HEG1の血圧制御における役割を解明し、その背後のシグナル伝達カスケードを特徴づけることを目的としました。
研究デザイン
本調査では、人間疫学、計算流体力学、実験的マウス遺伝学を統合した多モーダルアプローチを用いて、HEG1シグナル伝達と血圧制御の因果関係を確立しました。
人間の研究には、複数のコホートにおけるフェノーム広範囲関連解析、主幹動脈でのせん断応力パターンの特性評価、動脈試料からの内皮細胞の単一細胞RNAシークエンス、独立した検証コホートからの高血圧および正常血圧被験者の血漿HEG1濃度測定が含まれました。
実験的研究では、Tie2プロモーター下でのCre-lox再組合せによって生成された内皮特異的なHeg1ノックアウトマウスを使用しました。血圧モニタリングは、意識のある拘束されていない動物に対するテレメトリで行われました。血管機能は、ワイヤマイオグラフィを用いて内皮依存性および内皮非依存性血管拡張を評価しました。C57BL/6JとApoeノックアウト(ApoeKO)の両背景で研究が行われ、脂質異常症との相互作用を検討しました。
メカニズム研究では、プロテオミクス、トランスクリプトミクス、ユビキチン化アッセイ、核-細胞質分画を用いて、HEG1相互作用タンパク質と下流効果子を同定しました。PHACTR1核移動の薬理学的阻害薬CCG-1423を用いて、治療の可能性をテストしました。
主要な知見
高血圧患者におけるHEG1の低下
3つの独立したコホートにおいて、高血圧患者の血漿HEG1濃度は正常血圧対照群と比較して有意に低かったです(p < 0.001)。この低下は、頸動脈と尺骨動脈の計算流体力学解析から得られた壁面せん断応力の推定値の低下と相関していました。単一細胞RNAシークエンスは、HEG1発現が主に内皮細胞に見られ、低せん断応力領域にさらされた血管での転写レベルの低下を確認しました。
Heg1欠損時の血圧上昇
内皮特異的なHeg1ノックアウトマウスは、同胞対照群と比較して持続的な収縮期血圧の上昇を示しました(平均差:+12 mmHg、95% CI:8-16 mmHg、p < 0.001)。血圧上昇は、特にApoeKO背景で顕著でした。ここで、Heg1欠損マウスは、人間の第2段階高血圧に相当する高血圧を発症しました。舒張期血圧と平均動脈圧も同様に上昇しました。
血管拡張能力の障害
ワイヤマイオグラフィは、Heg1欠損大動脈と腸間膜抵抗動脈における内皮依存性血管拡張の著しい障害を示しました。アセチルコリンに対する量反応曲線は右にシフトし、最大反応が約60%低下しました(p < 0.001)。ナトリウムニトロプルシドによる内皮非依存性血管拡張は保たれており、特定の内皮メカニズムを確認しました。C57BL/6Jマウスでは基線で観察され、ApoeKO動物では高脂肪食で悪化しました。
分子メカニズム:HEG1-PHACTR1-eNOS軸
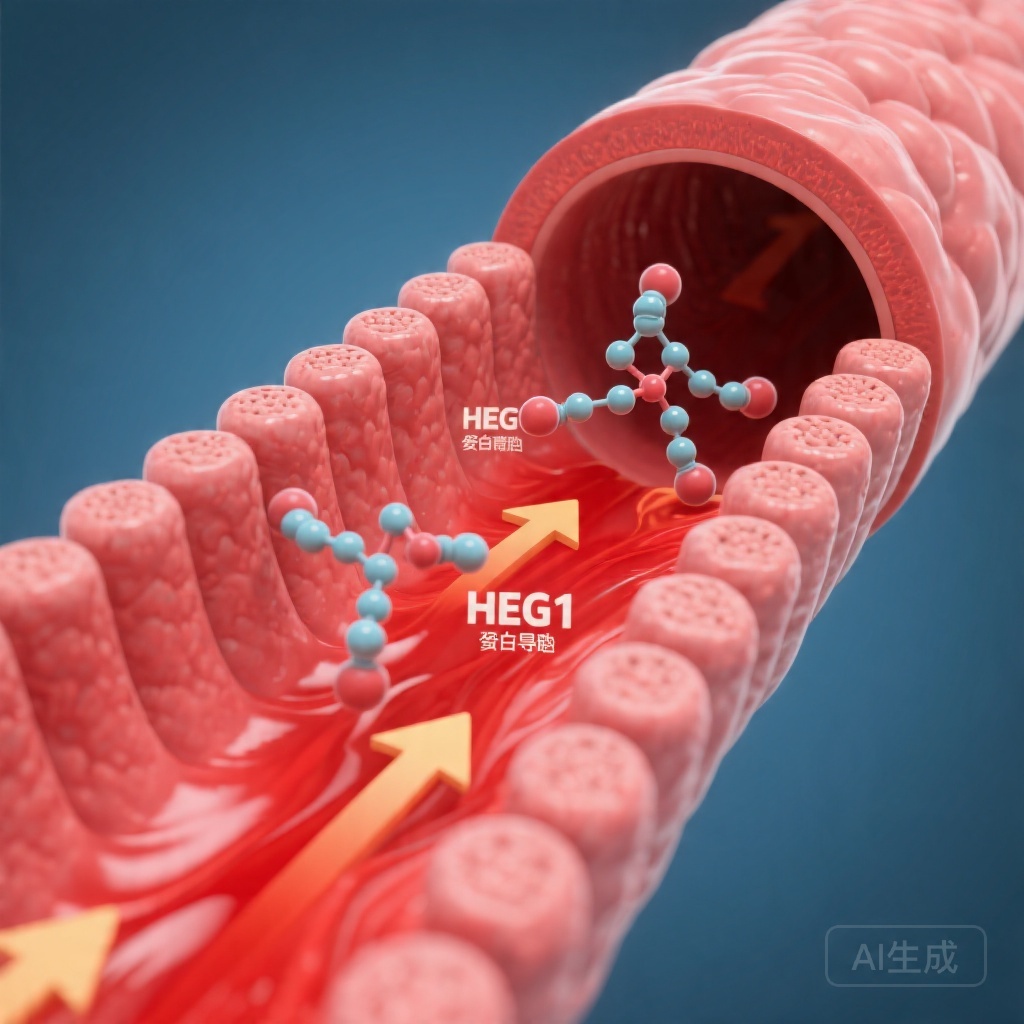
プロテオミクス解析では、PHACTR1(リン酸化酵素およびアクチンレギュレータ1)がHEG1によって相互補助的に制御される結合パートナーとして同定されました。HEG1は、Cullin-RINGユビキチンリガーゼ複合体のサcaffoldタンパク質であるCUL3と物理的に相互作用し、CUL3介在性ユビキチン化とプロテアソーム分解を促進しました。HEG1の欠如により、PHACTR1が蓄積し、核移動を起こしました。
核内では、PHACTR1はeNOS(内皮一酸化窒素合成酵素)のSP1依存性転写を抑制しました。クロマチン免疫沈降法は、Heg1欠損内皮細胞におけるeNOSプロモーターへのSP1結合の低下を確認しました。それに応じて、培養液と大動脈組織中のNO代謝物のレベルが大幅に低下しました。
薬理学的救済
PHACTR1核移動の阻害剤であるCCG-1423の投与は、Heg1欠損マウスにおける血圧上昇を予防し、内皮依存性血管拡張を回復させました。4週間の飲水投与により、核内PHACTR1蓄積が減少し、eNOS発現が野生型レベルに回復し、血圧が正規化されました。収縮期機能や腎機能パラメータには影響しませんでした。
主要なメカニズム経路
正常なせん断応力 → HEG1発現と放出 → CUL3介在性PHACTR1分解 → 核内PHACTR1の低レベル → SP1活性の抑制解除 → eNOS転写 → NO産生 → 血管拡張 → 正常な血圧
高血圧 ↓ せん断応力 → HEG1のダウンレギュレーション → PHACTR1の蓄積 → 核移動 → SP1の抑制 → eNOSの抑制 → NOの減少 → 血管拡張の障害 → 高血圧
専門家コメント
これらの知見は、HEG1が血液力学的力と内皮NO産生を結ぶ重要な機械感受性調節因子であることを確立しています。せん断応力とPHACTR1核移動の逆相関関係は、HEG1介在性ユビキチン化によって制御され、低または振動せん断応力が内皮機能不全と高血圧を引き起こす傾向がある分子的説明を提供しています。
遺伝子高血圧モデルでのPHACTR1核輸入の薬理学的阻害が正常な血圧を回復することを示す観察は、この経路が治療標的として利用可能であることを示唆しています。CCG-1423は、現時点で臨床承認されていませんが、新しいクラスの降圧薬の概念証明を表しています。
いくつかの制限点が考慮されるべきです。まず、人間の研究は相関的なものであり、遺伝子マウスモデルは翻訳的重要性を支持していますが、人間での介入研究が必要です。次に、ApoeKO背景は動脈硬化疾患に関連していますが、代謝的な混雑要因を導入します。さらに、PHACTR1阻害の長期安全性は、脳や腎臓を含む他の組織での役割を考えると確立されていません。
HEG1がCUL3とPHACTR1を結びつけるスキャフォールドとして機能することを示すメカニズム的洞察は、内皮メカノトランスダクションの理解を拡大しています。HEG1ベースの治療法が既存の降圧戦略を補完できるかどうか、特に難治性高血圧患者では、調査する必要があります。
結論
本研究は、内皮HEG1を中心に、せん断応力応答性シグナル伝達軸を特定し、PHACTR1ユビキチン化、核移動、下流eNOS発現の制御を通じて血圧を制御することを明らかにしました。高血圧患者における血漿HEG1の低下は、内皮メカノセンシングの低下を反映し、血流依存性血管拡張の障害のバイオマーカーとして機能する可能性があります。
せん断応力からHEG1、CUL3、PHACTR1、SP1を経てeNOS転写に至るメカニズムのカスケードは、血管生物学と全身的な血圧制御を結ぶこれまで認識されていなかった経路を表しています。PHACTR1核輸入のCCG-1423介在性阻害は、内皮機能不全に関連した高血圧の潜在的な治療戦略を提供します。
今後の方向性には、HEG1模倣ペプチドや小分子活性化剤の開発、血漿HEG1の予後バイオマーカーとしての検証、文書化された内皮機能不全を持つ高血圧集団でのPHACTR1核輸入阻害剤の臨床試験が含まれます。
資金源
本研究は、中国国家自然科学基金、中国医学科学院、欧州研究評議会からの助成金により支援されました。著者は利益相反を宣言していません。
参考文献
Wu W, Liu J, Chen X, Zhu P, Xu J, Yue J, Liu X, Fang J, Chen X, Pi J, Zheng L, Zhang Q, Zhang L, Schneider CV, Schneider KM, Trautwein C, Gao P, Reilly MP, Zhang Y, Zheng X, Liu J. Shear stress-induced endothelial HEG1 signalling regulates vascular tone and blood pressure. Eur Heart J. 2026;47(14):1721-1737. PMID: 40986512.