Highlights
Shear stress-induced endothelial HEG1 signalling represents a novel regulatory pathway linking blood flow dynamics to systemic blood pressure control. Plasma HEG1 levels are significantly reduced in hypertensive subjects due to diminished wall shear stress on the vascular endothelium. Endothelial-specific Heg1 deletion in mice results in elevated blood pressure, impaired endothelium-dependent vasodilation, and disrupted NO production. The mechanism involves HEG1-mediated regulation of PHACTR1 degradation, which controls SP1-dependent eNOS transcription.
Background
Hypertension remains a leading modifiable risk factor for cardiovascular disease, stroke, and renal dysfunction worldwide. Despite the availability of multiple antihypertensive agents, a substantial proportion of patients fail to achieve adequate blood pressure control, underscoring the need for novel therapeutic targets. Endothelial cells line the entire vascular system and serve as critical mechanosensors that translate hemodynamic forces, particularly wall shear stress, into biochemical signals governing vascular tone and blood pressure homeostasis.
Flow shear stress, the frictional force exerted by blood flow on the endothelial surface, stimulates vasodilation through mechanisms that remain incompletely understood. Impairment of this mechanotransduction pathway contributes to endothelial dysfunction, a hallmark of hypertension and atherosclerotic cardiovascular disease. Understanding the molecular intermediates that connect shear stress sensing to nitric oxide (NO) production may reveal actionable targets for blood pressure management.
Heart development protein with EGF-like domain 1 (HEG1) is an endothelial-derived membrane protein that has previously been associated with flow dynamics and cardiovascular phenotypes. Epidemiological data suggest an inverse relationship between HEG1 expression and cardiovascular risk factors, but the mechanistic basis for this association has not been defined. The present study aimed to elucidate the role of endothelial HEG1 in blood pressure regulation and to characterize the underlying signalling cascades.
Study Design
The investigation employed a multi-modal approach integrating human epidemiology, computational modelling, and experimental murine genetics to establish causality between HEG1 signalling and blood pressure control.
Human studies included phenome-wide association analysis across multiple cohorts, computational fluid dynamics to characterize shear stress patterns in conduit arteries, single-cell RNA sequencing of endothelial cells from arterial specimens, and measurement of plasma HEG1 concentrations in hypertensive and normotensive subjects from independent validation cohorts.
Experimental studies utilized endothelial-specific Heg1 knockout mice generated through Cre-lox recombination under the Tie2 promoter. Blood pressure monitoring was performed using telemetry in conscious, unrestrained animals. Vascular function was assessed through wire myography to evaluate endothelium-dependent and endothelium-independent vasodilation. Studies were conducted in both C57BL/6J and Apoe-knockout (ApoeKO) backgrounds to examine interactions with dyslipidaemia.
Mechanistic investigations employed proteomics, transcriptomics, ubiquitination assays, and nuclear-cytoplasmic fractionation to identify HEG1-interacting proteins and downstream effectors. Pharmacological inhibition of PHACTR1 nuclear translocation using CCG-1423 tested therapeutic potential.
Key Findings
Reduced HEG1 in Human Hypertension
Plasma HEG1 concentrations were significantly lower in hypertensive subjects compared with normotensive controls across three independent cohorts (p < 0.001). This reduction correlated with decreased wall shear stress estimates derived from computational fluid dynamics analysis of carotid and brachial arteries. Single-cell RNA sequencing confirmed HEG1 expression predominantly in endothelial cells, with reduced transcript levels in vessels exposed to low shear stress regions.
Blood Pressure Elevation Upon Heg1 Deletion
Endothelial-specific Heg1 knockout mice exhibited sustained elevation of systolic blood pressure compared with littermate controls (mean difference: +12 mmHg, 95% CI: 8-16 mmHg, p < 0.001). The blood pressure elevation was particularly pronounced in the ApoeKO background, where Heg1-deficient mice developed hypertension equivalent to human stage 2 hypertension. Diastolic blood pressure and mean arterial pressure were similarly elevated.
Impaired Vasodilatory Capacity
Wire myography revealed severe impairment of endothelium-dependent vasodilation in Heg1-deficient aortas and mesenteric resistance arteries. Dose-response curves to acetylcholine showed rightward shifts with maximal response reduced by approximately 60% (p < 0.001). Endothelium-independent vasodilation to sodium nitroprusside remained intact, confirming a specific endothelial mechanism. The impairment was observed at baseline in C57BL/6J mice and was exacerbated by high-fat diet in ApoeKO animals.
Molecular Mechanism: HEG1-PHACTR1-eNOS Axis
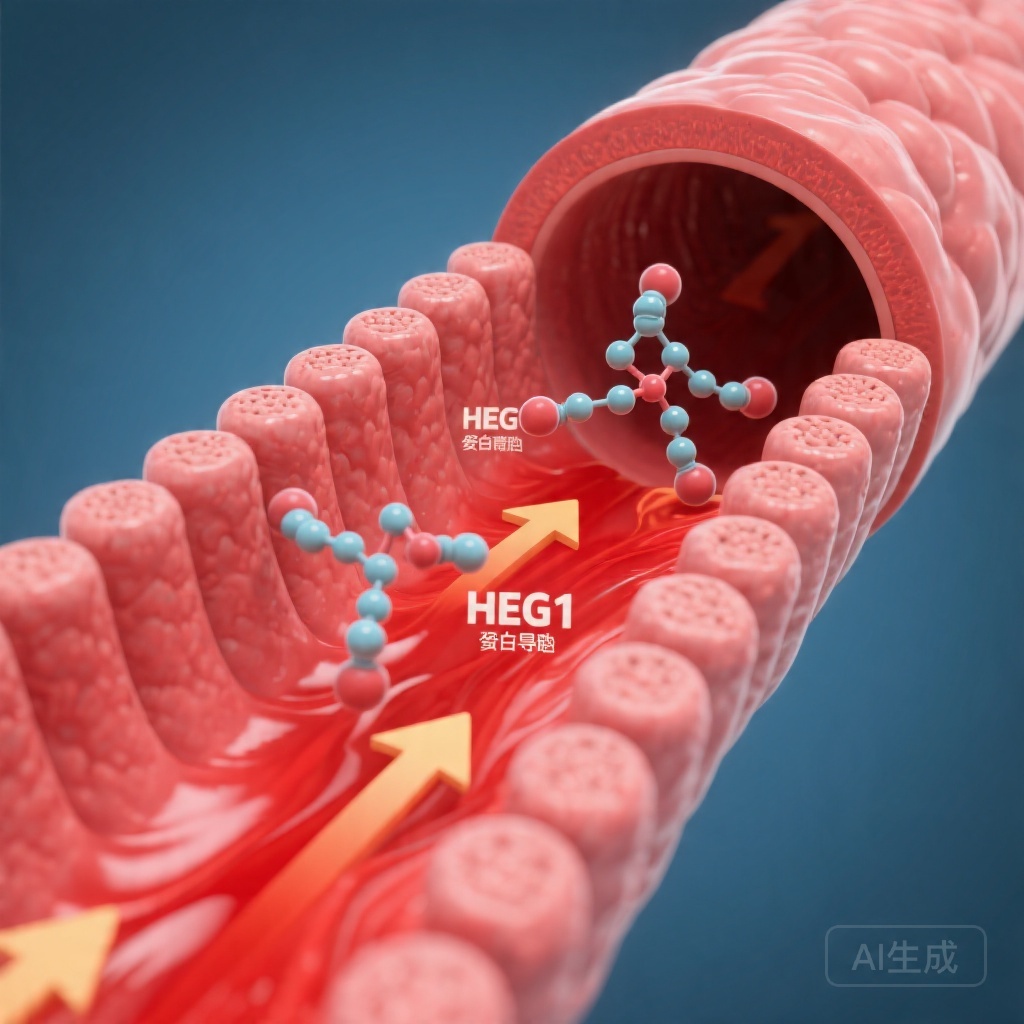
Proteomic analysis identified PHACTR1 (phosphatase and actin regulator 1) as a binding partner whose levels were reciprocally regulated by HEG1. HEG1 physically interacted with CUL3, a scaffold protein of the Cullin-RING E3 ubiquitin ligase complex, promoting CUL3-mediated ubiquitination and proteasomal degradation of PHACTR1. In the absence of HEG1, PHACTR1 accumulated and underwent nuclear translocation.
Within the nucleus, PHACTR1 suppressed SP1-mediated transcription of eNOS (endothelial nitric oxide synthase), the rate-limiting enzyme for NO production. Chromatin immunoprecipitation confirmed reduced SP1 binding to the eNOS promoter in Heg1-deficient endothelial cells. Accordingly, NO metabolite levels in conditioned media and aortic tissues were substantially reduced.
Pharmacological Rescue
Treatment with CCG-1423, a small molecule inhibitor of PHACTR1 nuclear localization, prevented blood pressure elevation and restored endothelium-dependent vasodilation in Heg1-deficient mice. Four-week administration via drinking water reduced nuclear PHACTR1 accumulation, restored eNOS expression to wild-type levels, and normalized blood pressure without affecting systolic function or renal parameters.
Key Mechanistic Pathway
Normal shear stress → HEG1 expression and release → CUL3-mediated PHACTR1 degradation → Low nuclear PHACTR1 → Uninhibited SP1 activity → eNOS transcription → NO production → Vasodilation → Normal blood pressure.
Hypertension ↓ shear stress → HEG1 downregulation → PHACTR1 accumulation → Nuclear translocation → SP1 inhibition → eNOS suppression → Reduced NO → Impaired vasodilation → Elevated blood pressure.
Expert Commentary
These findings establish HEG1 as a critical mechanosensitive regulator linking hemodynamic forces to endothelial NO production. The inverse relationship between shear stress and PHACTR1 nuclear localization, controlled by HEG1-mediated ubiquitination, provides a compelling molecular explanation for how regions of low or oscillatory shear stress predispose to endothelial dysfunction and hypertension.
The observation that pharmacological inhibition of PHACTR1 nuclear import restores normal blood pressure in genetic hypertension models suggests this pathway may be amenable to therapeutic targeting. CCG-1423, though not currently clinically approved, represents a proof-of-concept for a novel class of antihypertensive agents.
Several limitations merit consideration. First, the human studies are correlative; while the translational significance is supported by the genetic mouse models, interventional studies in humans are needed. Second, the ApoeKO background, while relevant to atherosclerotic disease, introduces confounding metabolic effects. Third, the long-term safety of PHACTR1 inhibition remains to be established, given the protein’s roles in other tissues including the brain and kidneys.
The mechanistic insight that HEG1 serves as a scaffold bringing together CUL3 and PHACTR1 expands our understanding of endothelial mechanotransduction. Whether HEG1-based therapies could complement existing antihypertensive strategies, particularly in patients with resistant hypertension, warrants investigation.
Conclusion
This study identifies a novel shear stress-responsive signalling axis centred on endothelial HEG1 that controls blood pressure through regulation of PHACTR1 ubiquitination, nuclear localization, and downstream eNOS expression. The reduction in plasma HEG1 observed in hypertensive patients reflects diminished endothelial mechanosensing and may serve as a biomarker of impaired flow-mediated vasodilation.
The mechanistic cascade from shear stress through HEG1, CUL3, PHACTR1, and SP1 to eNOS transcription represents a previously unrecognized pathway linking vascular biology to systemic blood pressure control. CCG-1423-mediated inhibition of PHACTR1 nuclear import offers a potential therapeutic strategy for hypertension associated with endothelial dysfunction.
Future directions include development of HEG1 mimetic peptides or small molecule activators, validation of plasma HEG1 as a prognostic biomarker, and clinical trials of PHACTR1 nuclear import inhibitors in hypertensive populations with documented endothelial dysfunction.
Funding
This study was supported by grants from the National Natural Science Foundation of China, the Chinese Academy of Medical Sciences, and the European Research Council. The authors declared no conflicts of interest.
References
Wu W, Liu J, Chen X, Zhu P, Xu J, Yue J, Liu X, Fang J, Chen X, Pi J, Zheng L, Zhang Q, Zhang L, Schneider CV, Schneider KM, Trautwein C, Gao P, Reilly MP, Zhang Y, Zheng X, Liu J. Shear stress-induced endothelial HEG1 signalling regulates vascular tone and blood pressure. Eur Heart J. 2026;47(14):1721-1737. PMID: 40986512.