Highlights
MAML1, a core Notch transcriptional coactivator, is implicated as a candidate gene in congenital heart disease, particularly ventricular septal defect.
A patient-derived MAML1 Q401K variant reproduced septal and valvular phenotypes in knock-in mice and CRISPR-edited human heart organoids.
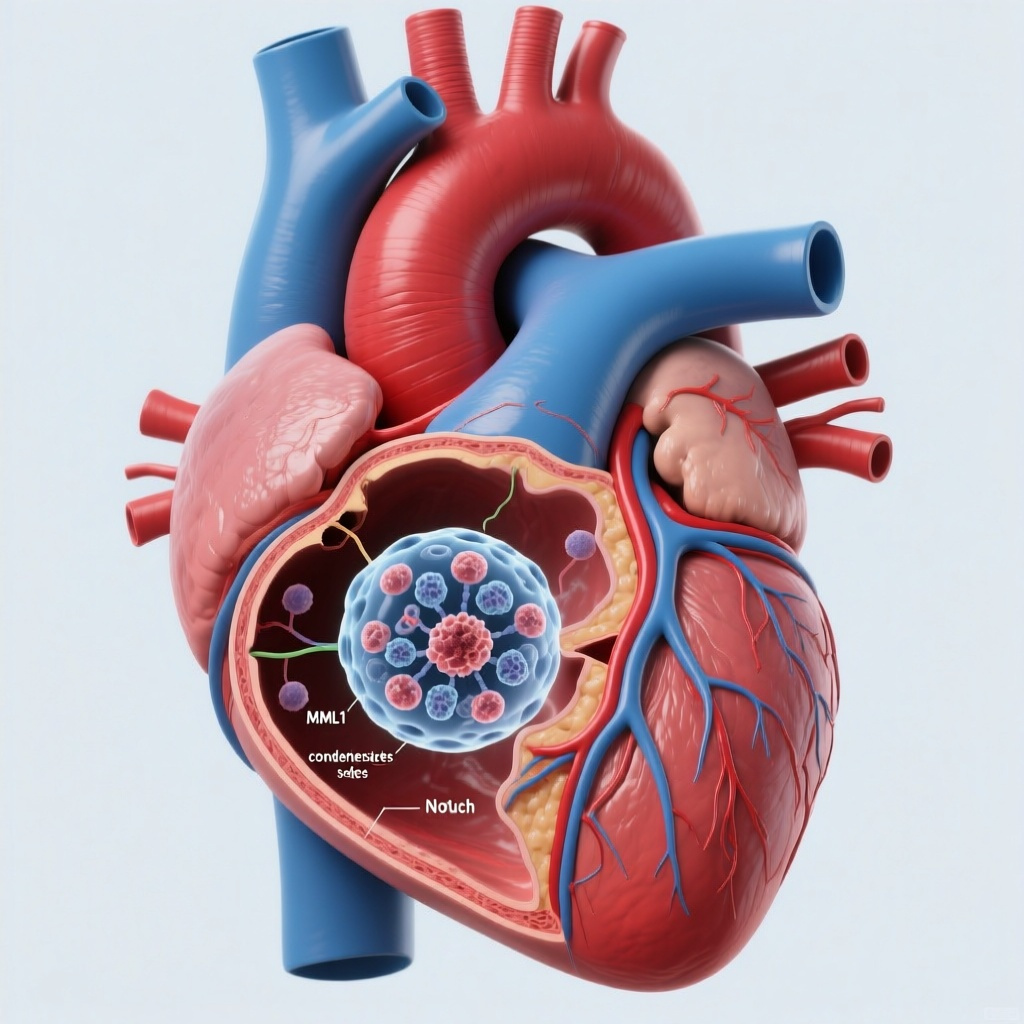
The study identifies liquid-liquid phase separation of MAML1 as a necessary biophysical mechanism for efficient Notch transcription in endocardial cells.
Both CHD-associated charge-altering variants and PKN2-mediated phosphorylation disrupted MAML1 condensates, converging on suppression of Notch-driven endocardial-to-mesenchymal transition.
Why This Study Matters
Congenital heart disease remains the most common birth defect and a major cause of infant morbidity and mortality. Although the genetic architecture of CHD is increasingly understood, many pathogenic mechanisms remain unresolved, especially those affecting cardiac morphogenesis at the level of transcriptional regulation. Notch signaling has long been recognized as central to endocardial cushion formation, valve development, and septation. Yet the precise ways in which transcriptional coactivators modulate Notch output in embryonic heart tissue have been less clear.
This study by Tan and colleagues, published in Circulation on June 5, 2026, addresses that gap by focusing on Mastermind-like 1 (MAML1), a known Notch transcriptional coactivator. The work goes beyond conventional genotype-phenotype association by linking rare patient variants to a specific biophysical defect: aberrant liquid-liquid phase separation (LLPS). In doing so, it places biomolecular condensate biology directly into the developmental cardiology framework.
Proposed Section Structure
The article is best interpreted through the following structure: clinical context and disease burden; study design and translational models; core phenotypic findings; mechanistic insights into MAML1 phase separation; implications for developmental cardiology and precision medicine; limitations and unanswered questions; and clinical-research takeaways.
Clinical Background
Notch signaling is indispensable during cardiac development. In the embryonic endocardium, it helps govern endocardial-to-mesenchymal transition, the process by which selected endocardial cells delaminate and populate the cardiac cushions that later form valves and septa. Disruption of this program has been associated with ventricular septal defects, outflow tract abnormalities, and valvular malformations. Human genetics has already implicated several components of the Notch pathway, including NOTCH1, JAG1, and DLL4, in congenital cardiovascular phenotypes.
MAML1 occupies a crucial position within this pathway. It acts as a transcriptional coactivator that binds the NOTCH intracellular domain and the DNA-binding factor RBPJ to promote target-gene expression. While its biochemical role in Notch transcription has been appreciated, whether altered MAML1 function contributes to human CHD and by what mechanism had not been established. The current study addresses both questions and identifies a mechanism that is not simply loss of protein abundance or failed DNA binding, but failure to form functional nuclear condensates.
Study Design and Experimental Platform
This was a multilevel translational investigation combining human genetics, mouse embryology, organoid modeling, and molecular biophysics.
Human genetic observation
The investigators identified rare missense MAML1 variants in a clinical cohort of patients with congenital heart disease, with an association particularly noted for ventricular septal defects. The centerpiece variant was Q401K, a patient-derived missense substitution.
In vivo mouse models
Two complementary murine approaches were used. First, a knock-in mouse carrying the Q401K variant tested whether the human allele was sufficient to produce disease-relevant cardiac phenotypes. Second, an endocardium-specific Maml1 knockout model assessed tissue specificity and clarified whether the pathogenic effect arose from loss of Maml1 activity in endocardial cells.
Human heart organoids
To strengthen translational relevance, the team generated CRISPR-edited human heart organoids harboring MAML1 deletion or patient-relevant mutation. This helped bridge the gap between mouse developmental biology and human cardiac morphogenesis.
Mechanistic assays
The authors used biochemical interaction assays, microscopy-based analyses of nuclear condensates, LLPS experiments, and mass spectrometry to examine the structural and regulatory basis of MAML1 function. They also identified an upstream kinase, PKN2, and mapped a relevant phosphorylation site at Ser314.
Key Findings
1. MAML1 variants are linked to congenital heart disease
The first important contribution is disease-gene nomination. Rare missense variants in MAML1 were found in patients with CHD, supporting the idea that MAML1 should be considered within the expanding set of candidate genes for structural congenital heart defects. The study is especially compelling because it does not stop at variant discovery. Instead, it functionally validates pathogenicity using both mammalian and human organoid systems.
2. The Q401K variant phenocopies ventricular septal defect biology
The Q401K knock-in mouse recapitulated key features relevant to ventricular septal defect pathogenesis. Histologic and functional analyses demonstrated septal and valvular abnormalities, aligning the model with clinically meaningful CHD phenotypes. Parallel findings in human heart organoids reinforced that the variant was not merely associated with disease but was sufficient to perturb developmental programs.
This cross-platform concordance is a notable strength. In developmental cardiology, phenotypic fidelity across species and model systems is often difficult to achieve. Here, the mouse and organoid results converged on a shared biological defect.
3. Endocardial MAML1 is required for proper septation and valvulogenesis
The endocardium-specific knockout experiments clarified tissue specificity. Loss of Maml1 in the endocardium caused defects similar to those seen with the Q401K variant, including abnormalities of septal and valvular development. These defects were mechanistically linked to impaired endocardial-to-mesenchymal transition, a process tightly governed by Notch signaling.
That observation is clinically important because it pinpoints the developmental compartment in which MAML1 function is most critical. Rather than causing broad embryonic failure, the pathogenic effect appears concentrated in endocardial Notch-dependent morphogenesis.
4. MAML1 requires liquid-liquid phase separation for effective Notch transcription
The study’s major conceptual advance is the demonstration that MAML1 activity depends on liquid-liquid phase separation. In the nucleus, MAML1 forms condensates that facilitate interaction with the NOTCH1 intracellular domain and support efficient transcriptional activation of downstream target genes. This reframes Notch regulation as not only a matter of canonical protein-protein assembly, but also one of mesoscale biophysical organization.
LLPS has become an important concept in transcription biology, particularly for proteins containing intrinsically disordered regions. This study identifies intrinsically disordered region 2 of MAML1 as a core phase-separating domain and places condensate integrity at the center of normal endocardial signaling.
5. Disease-associated variants disrupt electrostatic properties of MAML1 condensates
The pathogenic variants, including Q401K, were characterized as charge-altering substitutions within intrinsically disordered region 2. These changes abrogated normal LLPS behavior, weakened nuclear condensate formation, and reduced Notch transcriptional output. The implication is elegant: a single amino acid substitution can shift the electrostatic landscape of a disordered protein region enough to impair condensate assembly and thereby alter developmental signaling.
This is a particularly strong mechanistic link because it connects genotype, protein biophysics, signal transduction, and organ-level malformation in one coherent chain.
6. PKN2 phosphorylation provides an upstream regulatory brake
The investigators also identified a regulatory pathway independent of inherited mutation. PKN2 phosphorylated MAML1 at Ser314, destabilizing MAML1 condensates and attenuating Notch transcriptional activity. This finding suggests that congenital malformation or developmental susceptibility may arise not only from structural MAML1 variants, but also from dysregulated posttranslational control of MAML1 phase behavior.
That expands the translational significance of the work. It raises the possibility that MAML1 condensates represent a regulatable signaling node, potentially vulnerable to environmental, metabolic, or kinase-mediated perturbation during embryogenesis.
Mechanistic Interpretation
The biology proposed by the authors is internally consistent and plausible. MAML1 is known to function as a scaffold within the canonical Notch transcriptional complex. Proteins with intrinsically disordered regions often rely on weak multivalent interactions to form dynamic condensates, which can concentrate cofactors and RNA polymerase machinery at active transcriptional sites. The present study suggests that MAML1 condensates serve precisely this purpose in endocardial nuclei.
When the electrostatic balance of these regions is altered by a missense variant such as Q401K, condensate assembly becomes defective. As a result, MAML1 interacts less efficiently with the NOTCH1 intracellular domain, downstream Notch targets are underexpressed, and endocardial-to-mesenchymal transition is impaired. PKN2 phosphorylation at Ser314 appears to act in a parallel fashion by destabilizing these condensates, again lowering transcriptional output.
For clinicians and translational scientists, the broader message is that some developmental heart defects may result from disordered nuclear organization rather than classic enzyme deficiency or receptor loss. This has implications for variant interpretation, especially when apparently subtle missense changes occur within intrinsically disordered regions that are often harder to classify by standard structural models.
Clinical and Translational Relevance
Several practical implications follow from this work.
First, MAML1 merits consideration in research-based CHD sequencing panels and in variant curation workflows, especially in patients with ventricular septal or valvular phenotypes and suspected Notch-pathway disease.
Second, the findings reinforce the primacy of endocardial Notch signaling in septation and valve formation, which may help organize future studies of genotype-phenotype correlations in nonsyndromic CHD.
Third, this study illustrates why functional assays are increasingly necessary in cardiovascular genetics. Variants in intrinsically disordered regions may not be interpretable through conventional protein structure prediction alone. Condensate behavior, charge distribution, and posttranslational modification can be disease-defining.
Fourth, the PKN2-MAML1 axis introduces a possible therapeutic concept, though this remains highly preliminary. If Notch suppression in some developmental contexts is mediated by aberrant condensate destabilization, then modulation of upstream kinases or condensate-supporting interactions could become a future strategy. At present, however, this is a mechanistic insight rather than a clinically actionable pathway.
Strengths of the Study
The study is notable for its breadth and coherence. It integrates patient-derived genetics with knock-in mice, tissue-specific knockout mice, human organoids, imaging of condensate biology, and phosphoproteomic-type mechanistic analysis. The use of both gain-of-dysfunction modeling through Q401K and tissue-restricted loss-of-function modeling through endocardial knockout is especially persuasive. So too is the triangulation across mouse and human systems.
Another strength is the specificity of the mechanism. The paper does not merely show reduced Notch activity; it identifies the structural region involved, the nature of the physicochemical defect, and an upstream kinase that modulates the same process.
Limitations and Open Questions
As with most high-impact mechanistic studies, some caution is warranted.
The abstract does not provide effect sizes, penetrance estimates, or confidence intervals for the human genetic association, so the epidemiologic strength of MAML1 as a recurrent CHD gene will need confirmation in larger independent cohorts.
Second, although organoids are a powerful translational model, they do not fully replicate blood flow, biomechanical forces, and spatial patterning of the developing human heart. Some phenotypes may be incompletely modeled.
Third, LLPS is a useful framework, but the field continues to debate how best to distinguish true phase separation from other forms of dynamic molecular clustering in living cells. The authors appear to have used multiple orthogonal assays, which strengthens the claim, but replication by independent groups will be important.
Fourth, the clinical consequences of PKN2-mediated MAML1 phosphorylation remain uncertain. It is not yet known whether this axis is altered in human CHD specimens, whether it is developmentally stage-specific, or whether it interacts with maternal or environmental exposures.
Finally, it remains to be determined whether MAML1-related CHD is largely limited to ventricular septal and valvular defects or contributes to a wider spectrum of conotruncal and left-sided lesions associated with abnormal Notch signaling.
Conclusion
Tan and colleagues provide compelling evidence that MAML1 is a candidate congenital heart disease gene and that its pathogenicity can arise through defective liquid-liquid phase separation in endocardial nuclei. By linking patient-derived missense variation to abnormal condensate formation, suppressed Notch signaling, and failed endocardial-to-mesenchymal transition, the study opens a new mechanistic chapter in developmental cardiology.
The central translational insight is that the electrostatic integrity of transcriptional condensates can be essential for normal heart morphogenesis. This work broadens how clinicians and scientists should think about CHD genetics: not only in terms of absent proteins or disrupted pathways, but also in terms of altered biophysical state. Future studies should define the population-level contribution of MAML1 variants, validate biomarkers of pathway dysfunction, and explore whether condensate-regulating mechanisms could eventually be targeted in developmental disease models.
Funding and Trial Registration
The abstract provided does not report funding details or a ClinicalTrials.gov registration number. As this is a mechanistic preclinical and translational study rather than an interventional clinical trial, trial registration may not be applicable.
Citation
Tan Z, Qi Y, Chen Y, Zhang L, Li C, Lei B, Xiao Z, Zhang J, Lu L, Wang H. Aberrant Phase Separation of Endothelial MAML1 Causes Congenital Heart Disease by Suppressing Notch Activity. Circulation. 2026-06-05. PMID: 42246060. URL: https://pubmed.ncbi.nlm.nih.gov/42246060/
Contextual References
MacGrogan D, Luxán G, de la Pompa JL. Notch signaling in cardiac development and disease. Curr Top Dev Biol. 2010;92:333-365.
Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270-274.
Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451(7181):943-948.
de Soysa TY, Ranade SS, Okawa S, et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature. 2019;572(7767):120-124.