Highlights
Single-cell transcriptomic profiling of rare human pancreatic tissue from critically ill patients with severe acute pancreatitis identified pancreatic microcirculatory failure as a central disease mechanism rather than a secondary epiphenomenon.
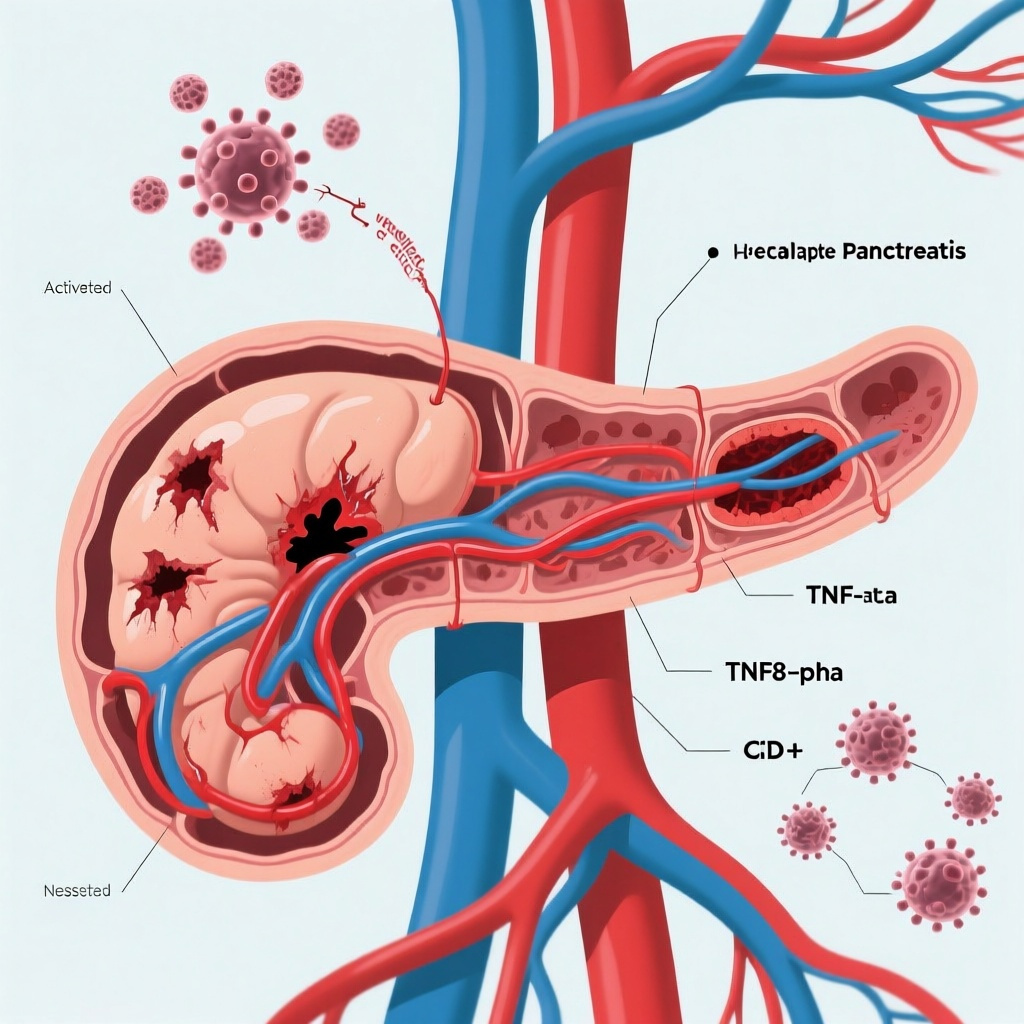
The study implicates a specific immune-endothelial circuit in which TNF-α induces endothelial UL16-binding proteins, activating NKG2D-positive CD8+ T cells and amplifying endothelial injury.
Clinical correlation in a larger patient cohort linked this TNF-α/ULBP/NKG2D axis to disease severity and microcirculatory dysfunction, strengthening the translational relevance of the mechanistic findings.
In murine and porcine models, inhibition of TNF-α or depletion of CD8+ T cells preserved microvascular integrity, reduced pancreatic injury, and improved survival, providing a preclinical rationale for therapeutic targeting.
Background and Clinical Context
Acute pancreatitis is one of the most common gastrointestinal causes of hospital admission worldwide. While most cases are self-limited, a substantial minority progress to moderately severe or severe disease, characterized by persistent organ failure, pancreatic necrosis, infected collections, and high mortality. Severe acute pancreatitis (SAP) remains a major challenge in gastroenterology, surgery, and critical care because treatment is still largely supportive. Despite decades of study, no targeted immune-modulating therapy has entered routine care.
The classical pathophysiologic framework of SAP emphasizes premature digestive enzyme activation, acinar cell injury, sterile inflammation, cytokine release, and later infectious complications. Yet one important feature has long been recognized but insufficiently mechanistically defined: pancreatic microcirculatory dysfunction. Reduced capillary perfusion, endothelial activation, leukocyte adhesion, thrombosis, and local ischemia are thought to worsen necrosis and propagate systemic inflammation. Human evidence has been difficult to obtain because pancreatic tissue from patients with SAP is rarely available, especially at a stage informative for primary disease mechanisms.
This study by Shi and colleagues addresses that gap by combining single-cell transcriptomics from human SAP tissue with clinical correlation and experimental validation. The result is a more precise model of disease progression, centered on immune-mediated endothelial injury and the possibility of interrupting that process with TNF-α inhibition.
Study Design and Methods
The investigators analyzed pancreatic tissue from 8 critically ill patients with SAP, generating what they describe as the first single-cell transcriptomic atlas of human SAP. This is an important design strength because human pancreatic data in severe disease are exceptionally scarce, and single-cell methods can resolve cell-specific states and interactions that bulk transcriptomic approaches cannot.
The human tissue analysis focused on two broad questions: which cellular compartments are altered in SAP, and which ligand-receptor communication pathways may account for progression of pancreatic injury. The authors evaluated shifts in cellular composition and inferred intercellular signaling networks, particularly those involving endothelial cells and immune infiltrates.
To assess clinical relevance, transcriptomic findings were integrated with a larger patient cohort of 153 individuals. Although the abstract does not provide granular cohort characteristics, this step appears to have been used to correlate the proposed molecular axis with disease severity and evidence of microcirculatory dysfunction.
Mechanistic validation was then pursued in both in vitro and in vivo systems. The in vivo models included murine and porcine SAP models, an important translational feature because porcine physiology can better approximate human vascular and inflammatory responses than rodent systems alone. Interventions included inhibition of TNF-α and targeting of CD8+ T cells, with assessment of microvascular integrity, tissue damage, and survival.
Key Findings
1. Human SAP tissue reveals microcirculatory failure as a core pathogenic process
The central conceptual advance of the study is that pancreatic microvascular injury is not merely a downstream marker of severe inflammation, but a critical driver of disease progression. Single-cell profiling identified endothelial perturbation as a prominent feature of SAP. Although the abstract does not provide the complete cellular atlas, the interpretation is clear: endothelial cells emerge as active participants in the disease process, not passive casualties of surrounding inflammation.
This matters clinically. Pancreatic perfusion abnormalities have long been associated with necrosis, organ failure, and adverse outcomes, but the field has lacked a convincing human cellular mechanism linking inflammatory mediators to endothelial damage in a way that could be therapeutically targeted. The present study places endothelial dysfunction near the center of SAP pathobiology.
2. CD8+ T cells are implicated as direct mediators of endothelial injury
The authors identify infiltrating CD8+ T cells as key effectors of pancreatic endothelial injury. This is notable because innate immune cells, especially neutrophils and macrophages, have traditionally received most of the attention in acute pancreatitis research. The study suggests that adaptive cytotoxic lymphocytes may play a more direct and damaging role in the pancreatic microenvironment than previously appreciated.
The proposed model is that activated CD8+ T cells recognize stressed endothelial cells and inflict cytotoxic damage, thereby worsening capillary leak, perfusion failure, and local ischemia. This immune-mediated endothelial attack provides a plausible explanation for how inflammation can be converted into sustained microvascular collapse and pancreatic tissue destruction.
3. TNF-α drives endothelial stress signaling through ULBP induction
Mechanistically, TNF-α was shown to upregulate UL16-binding proteins (ULBPs) on endothelial cells. ULBPs are ligands for the activating receptor NKG2D, which is expressed on cytotoxic lymphocytes including subsets of CD8+ T cells. In this framework, TNF-α does more than amplify generalized inflammation: it functionally “primes” endothelial cells for immune attack by increasing expression of stress ligands that are recognized by NKG2D-positive T cells.
This is an elegant mechanistic bridge between cytokine biology and vascular injury. TNF-α has long been implicated in systemic inflammatory states, but broad cytokine elevation alone does not explain organ-selective injury. By identifying an endothelial stress-ligand pathway, the study narrows the mechanism to a defined immune-recognition axis.
4. The TNF-α/ULBP/NKG2D axis forms a feed-forward loop
One of the most compelling features of the proposed mechanism is the presence of a feed-forward circuit. TNF-α increases endothelial ULBP expression; ULBPs engage NKG2D on infiltrating CD8+ T cells; activated cytotoxic T cells injure endothelium; worsening endothelial injury likely promotes further inflammatory activation and microcirculatory dysfunction. Such self-reinforcing loops are often what distinguish transient inflammation from fulminant organ injury.
From a translational standpoint, feed-forward loops are attractive therapeutic targets because interruption at one node can potentially dampen the entire network. In this study, TNF-α appears to be an upstream and actionable node.
5. Clinical correlation supports biological relevance
The study did not stop at discovery biology. In a patient cohort of 153 individuals, the TNF-α/ULBP/NKG2D axis correlated with disease severity and microcirculatory dysfunction. Even without detailed effect sizes in the abstract, this clinical association is important because it reduces the risk that the transcriptomic findings are epiphenomena confined to end-stage tissue sampling.
Correlation with severity also raises the possibility that this pathway could have biomarker utility. If circulating or tissue-surrogate markers of TNF-α signaling, endothelial stress, or NKG2D activation can be validated, clinicians may eventually be able to identify patients most likely to benefit from targeted intervention.
6. Preclinical intervention preserved microvascular integrity and improved survival
The most clinically provocative finding is that TNF-α inhibition or CD8+ T-cell targeting improved outcomes in animal models. According to the abstract, these interventions preserved microvascular integrity, reduced tissue damage, and improved survival. This shifts the study from descriptive mechanistic work toward therapeutic proof of concept.
Importantly, the observed benefit was not limited to histologic injury. Improvement in survival, especially when demonstrated across more than one species, strengthens the argument that the identified pathway is functionally important. The inclusion of a porcine model further enhances translational credibility, as large-animal validation is often lacking in preclinical pancreatitis research.
Why These Findings Matter Clinically
The major unmet need in SAP is the absence of disease-modifying therapy. Current management still relies on aggressive fluid resuscitation, pain control, enteral nutrition, organ support, treatment of complications, and selective intervention for infected necrosis or biliary obstruction. None of these approaches specifically interrupts the molecular pathways that convert pancreatic injury into sustained organ failure.
This study suggests that TNF-α blockade could be revisited in a more biologically informed way. Historically, anti-cytokine therapy in acute inflammatory syndromes has often produced disappointing or inconsistent results, partly because patient populations were heterogeneous and mechanistic targets were too broadly conceived. Here, the authors provide a more precise rationale: TNF-α may be pathogenic not simply because it is elevated, but because it induces endothelial stress ligands that trigger CD8+ T-cell cytotoxicity and microvascular collapse.
If validated, this could reshape the therapeutic conversation in SAP. Rather than viewing anti-TNF treatment as generic immunosuppression, clinicians may begin to consider it as a strategy to preserve pancreatic microcirculation and prevent irreversible necrotizing injury in selected high-risk patients.
Expert Commentary and Critical Appraisal
This work has several clear strengths. First, the use of actual human pancreatic tissue is a major advance in a field where mechanistic inference has often depended heavily on animal models. Second, single-cell resolution allows identification of cell-type-specific interactions that would otherwise be obscured. Third, the investigators linked discovery findings to a larger clinical cohort and then to interventional experiments in both murine and porcine systems, creating a coherent translational arc from bedside to bench and back.
At the same time, several caveats deserve attention. The human tissue sample size was necessarily small at 8 patients, which is understandable but still limits assessment of heterogeneity. The timing of tissue acquisition in relation to symptom onset, organ failure, infection status, and supportive therapies could all influence the observed cellular states. SAP is not a single biological entity; biliary, hypertriglyceridemic, alcoholic, post-procedural, and idiopathic pancreatitis may have overlapping but nonidentical immune programs.
Another important question is therapeutic window. In clinical practice, patients often present after substantial tissue injury has already occurred. It remains uncertain whether TNF-α blockade would need to be administered very early to preserve endothelial function, or whether benefit persists once necrosis and organ dysfunction are established. Similarly, safety will be a major issue. Anti-TNF therapy could theoretically increase the risk of infection in a syndrome already complicated by infected necrosis, bacteremia, ventilator-associated pneumonia, and invasive procedures.
The study also raises the possibility that not all patients with SAP should be treated identically. A biomarker-enriched strategy may be necessary, targeting those with a strong TNF-α/ULBP/NKG2D signature or early evidence of pancreatic microcirculatory failure. This precision-medicine angle is promising but not yet clinically operational.
Finally, enthusiasm should be tempered until interventional human data become available. Acute pancreatitis has a long history of plausible biological targets that failed in trials. The present work is stronger than much prior preclinical literature because it begins with human tissue, but it still does not establish clinical efficacy.
Relationship to Existing Knowledge
Current international guidance emphasizes early supportive care and complication management, reflecting the lack of validated targeted therapy in acute pancreatitis. The revised Atlanta classification formalized the prognostic importance of persistent organ failure and local complications, while modern reviews have highlighted endothelial injury, coagulation disturbances, and dysregulated immunity as contributors to severe disease. What has been missing is a human, cell-resolved explanation linking cytokine signaling to pancreatic vascular collapse.
This study fills part of that gap. It also expands the immune framework of pancreatitis beyond innate inflammation by identifying a role for CD8+ T-cell cytotoxicity. That observation may stimulate broader investigation into adaptive immune pathways in pancreatic injury, including whether similar mechanisms operate in infected necrosis, recurrent acute pancreatitis, or progression to chronic pancreatic damage.
Implications for Research and Practice
For clinicians, the immediate implication is not a change in standard care but a change in how SAP may be conceptualized. Pancreatic microcirculation should be viewed as a therapeutic target rather than only a marker of severity. This may also increase interest in bedside tools that capture perfusion failure, including advanced imaging, endothelial biomarkers, and microvascular monitoring strategies.
For researchers, several next steps are clear. Prospective validation of the TNF-α/ULBP/NKG2D pathway in independent patient cohorts is needed, ideally with serial sampling to define temporal dynamics. Biomarker development will be essential if early-phase clinical trials are to enrich for likely responders. Interventional studies should clarify treatment timing, dosing, and safety, especially with respect to secondary infection. It may also be worth comparing direct TNF-α inhibition with more selective targeting of NKG2D signaling or endothelial stress responses.
For drug development, the study is attractive because TNF-α is already a well-known therapeutic target with existing biologic agents. Repurposing an established drug class may shorten translational timelines, although acute-care dosing, route, and patient selection will require careful redesign for SAP.
Conclusion
Shi and colleagues provide a notable advance in the mechanistic understanding of severe acute pancreatitis. By generating a single-cell atlas of human SAP and integrating it with clinical and experimental validation, they identify pancreatic microcirculatory failure as a central driver of disease progression. The proposed mechanism—TNF-α induction of endothelial ULBPs, activation of NKG2D-positive CD8+ T cells, and feed-forward endothelial injury—offers both biological coherence and therapeutic plausibility.
The translational message is clear: TNF-α is not just an inflammatory bystander in SAP, but a potential upstream driver of immune-endothelial damage. Whether this insight will translate into effective treatment for patients remains to be tested, but the study provides one of the strongest mechanistic rationales to date for targeted therapy in severe acute pancreatitis.
Funding and ClinicalTrials.gov
Funding information was not provided in the abstract or PubMed entry supplied. No ClinicalTrials.gov registration was reported for this translational tissue and preclinical study.
References
1. Shi L, Li W, Chen D, Liu B, Huang Z, Jin C, Ma L, Liu Q, Dong B, Pan Z, Du L, Hou L, Chen M, Xie J, Bai R, Gu H, Wang D, Yu X, Shen B, Qian J, Yu H. TNF-α drives pancreatic microcirculatory dysfunction via CD8+ T cell-mediated endothelial injury in severe acute pancreatitis. Gut. 2026-06-03. PMID: 42236225.
2. Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, Tsiotos GG, Vege SS; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62(1):102-111. PMID: 23100216.
3. Boxhoorn L, Voermans RP, Bouwense SA, Bruno MJ, Verdonk RC, Boermeester MA, van Santvoort HC, Besselink MG. Acute pancreatitis. Lancet. 2020;396(10252):726-734. PMID: 32891214.
4. Crockett SD, Wani S, Gardner TB, Falck-Ytter Y, Barkun AN; American Gastroenterological Association Institute Clinical Guidelines Committee. American Gastroenterological Association Institute Guideline on initial management of acute pancreatitis. Gastroenterology. 2018;154(4):1096-1101. PMID: 29409760.
5. Leppäniemi A, Tolonen M, Tarasconi A, Segovia-Lohse H, Gamberini E, Kirkpatrick AW, Ball CG, Parry N, Sartelli M, Wolbrink D, van Goor H, Baiocchi G, Ansaloni L, Biffl W, Coccolini F, Di Saverio S, Kluger Y, Moore E, Catena F. 2019 WSES guidelines for the management of severe acute pancreatitis. World J Emerg Surg. 2019;14:27. PMID: 31170955.

