Highlights
– Diabetes increases myocardial burden of neutrophil extracellular traps (NETs) and promotes neutrophil inflammasome activation in humans and mice.
– PAD4 deficiency in mice prevents glucose-triggered NETosis and ASC speck formation, reducing IL‑1β, VWF, neutrophil infiltration, fibrosis and preserving cardiac and renal function after experimental diabetes.
– The study links PAD4-dependent NET formation and inflammasome signaling as mechanistic mediators of diabetic cardiorenal injury and identifies PAD4/NETs as potential therapeutic targets.
Background: disease burden and unmet need
Both cardiovascular disease and chronic kidney disease are major complications of diabetes and account for substantial morbidity, mortality, and healthcare costs worldwide. Diabetic cardiomyopathy (DCM) and diabetic kidney disease (DKD) share common pathophysiologic features—chronic low-grade inflammation, microvascular dysfunction, extracellular matrix remodelling, and progressive fibrosis—that contribute to heart failure and progressive renal impairment. Despite advances in glucose-lowering therapies and recent cardiorenal-protective drugs (e.g., SGLT2 inhibitors), a significant residual risk remains and the cellular mediators linking hyperglycaemia to organ fibrosis are incompletely defined.
Study design
The referenced study (Schommer et al., Eur Heart J. 2025) examined the role of peptidylarginine deiminase 4 (PAD4), neutrophil extracellular traps (NETs), and inflammasome activation in human and experimental diabetes-associated cardiorenal injury.
Key elements:
- Human component: endomyocardial biopsies (EMB) from 20 heart failure patients with and without diabetes were analysed for NET burden.
- Mouse studies: wild-type (WT) and PAD4−/− mice received streptozotocin (STZ) to induce experimental diabetes. Longitudinal assessments included blood glucose, body weight, cardiac function, exercise tolerance, and pulmonary oedema.
- Cellular assays: human and mouse neutrophils were exposed to high glucose to evaluate NETosis and inflammasome activation (ASC speck formation). Measurements included confocal microscopy, ELISA for IL‑1β and von Willebrand factor (VWF), flow cytometry, and histologic fibrosis assessment (Sirius Red/Fast Green). Renal function was assessed by albuminuria and histology.
Key findings
The study reports a coherent set of observations linking hyperglycaemia to neutrophil-driven inflammation and organ fibrosis:
1. Increased myocardial NET burden in patients with diabetes
Endomyocardial biopsies from heart failure patients with diabetes showed an increased burden of NETs compared with those without diabetes, supporting clinical relevance of NETs in diabetic cardiac injury.
2. High glucose activates the neutrophil inflammasome
Exposure of human neutrophils to high-glucose conditions triggered inflammasome activation as evidenced by ASC speck formation; this provides a mechanistic link between hyperglycaemia and innate immune activation.
3. PAD4 is required for diabetes-triggered NETosis and inflammasome activation in mice
Both WT and PAD4−/− mice developed hyperglycaemia and weight loss after STZ, indicating comparable glycaemic injury. However, only WT neutrophils showed increased NETosis and ASC speck formation following diabetes. PAD4−/− neutrophils were resistant to high-glucose-induced NET formation and inflammasome signalling—implicating PAD4 as a critical mediator.
4. PAD4 deficiency protects against diabetes-associated cardiac dysfunction and systemic inflammation
Only diabetic WT mice exhibited elevated circulating IL‑1β and VWF, impaired cardiac function, reduced exercise tolerance and pulmonary oedema. PAD4−/− diabetic mice were protected from these adverse phenotypes despite comparable hyperglycaemia. These findings suggest PAD4-mediated NETosis amplifies systemic inflammation and endothelial perturbation in diabetes.
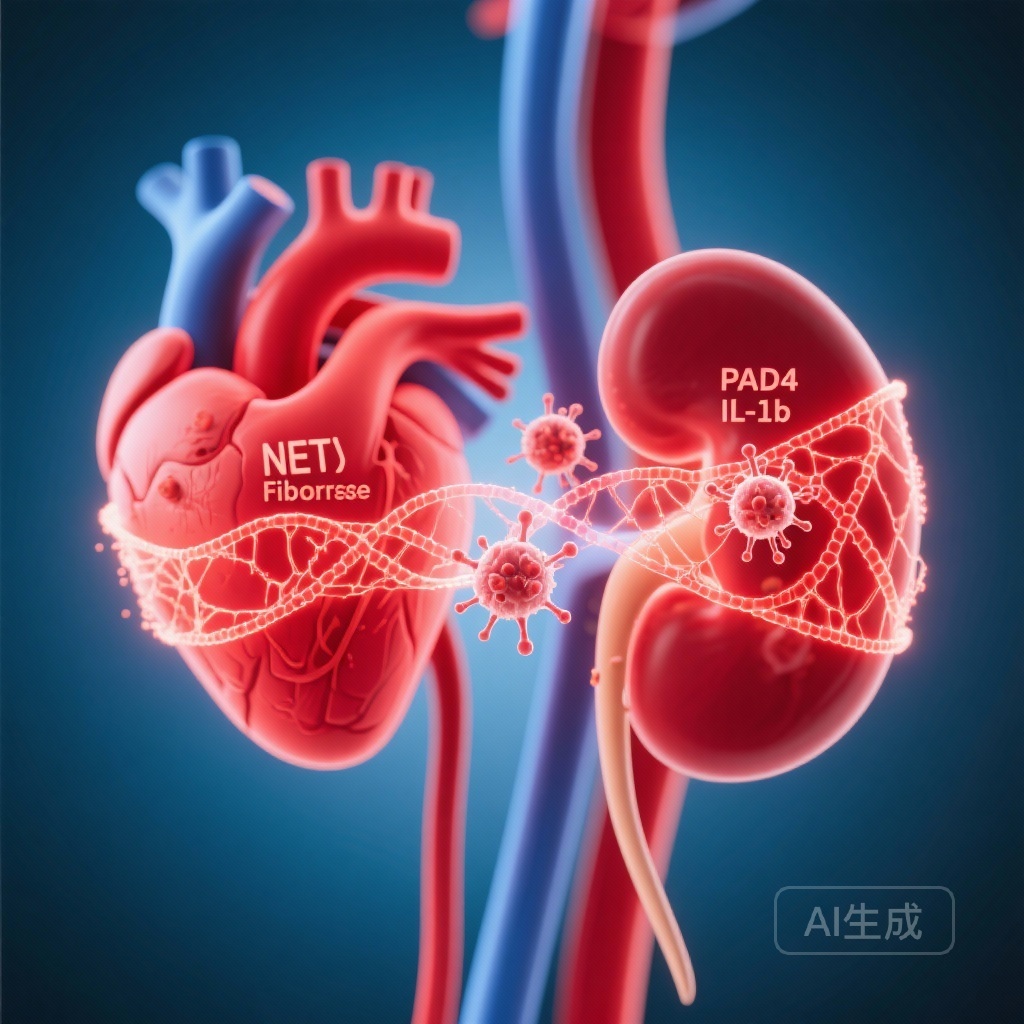
5. PAD4 drives cardiac and renal inflammation and fibrosis
Histologic analysis showed greater cardiac and renal fibrosis, neutrophil infiltration, NET deposition, and increased transforming growth factor‑β1 (TGF‑β1) levels in diabetic WT mice. Functionally, WT mice developed albuminuria and renal fibrosis consistent with DKD, whereas PAD4−/− diabetic mice preserved renal function and had reduced fibrotic changes.
6. Integrative interpretation
Taken together, the data support a model in which hyperglycaemia stimulates neutrophil inflammasome activation and PAD4-dependent NETosis; NETs and inflammasome-derived IL‑1β amplify endothelial activation (reflected by VWF), recruit more neutrophils, and promote TGF‑β1–driven fibrotic remodelling in heart and kidney, ultimately leading to dysfunction and heart failure.
Mechanistic and translational insights
This study provides both mechanistic depth and translational relevance. Important mechanistic nodes include:
- PAD4-mediated histone citrullination: PAD4 citrullinates histones, promoting chromatin decondensation and NET release—a central step in NETosis.
- Neutrophil inflammasome: ASC speck formation indicates activation of inflammasome complexes (e.g., NLRP3) in neutrophils, culminating in caspase‑1 activation and IL‑1β release; IL‑1β is a potent pro‑fibrotic and pro‑inflammatory cytokine.
- Cross-talk with vascular endothelium and profibrotic pathways: NET components and IL‑1β induce endothelial perturbation (VWF release) and TGF‑β1 expression, promoting collagen deposition and organ fibrosis.
These pathways are biologically plausible and consistent with prior literature linking NETs to sterile inflammation, thrombosis, and tissue injury (Brinkmann et al., 2004; Papayannopoulos, 2018) and implicating inflammasome/IL‑1β signalling in metabolic inflammation (Vandanmagsar et al., 2011).
Clinical and therapeutic implications
Key translational opportunities arise from interruption of the PAD4–NET–inflammasome axis:
- PAD4 inhibition: Genetic PAD4 deficiency was protective in mice; selective PAD4 inhibitors (under preclinical/early clinical development) may reduce NETosis and downstream fibrotic consequences in diabetes.
- NET-targeting strategies: DNase-based therapies that degrade extracellular DNA or agents that block NET formation could be repurposed or tested for cardiorenal protection.
- Inflammasome/IL‑1β blockade: Clinical trials (e.g., CANTOS, Ridker et al.) support IL‑1β inhibition for cardiovascular risk reduction; this study suggests IL‑1β blockade might also modulate cardiorenal fibrotic responses in diabetes, though organ-specific effects require study.
- Biomarker development: Circulating markers of NETosis (cell-free DNA, citrullinated histone H3) and inflammasome activation could serve as mechanistic biomarkers to identify high-risk patients or monitor response to targeted interventions.
Study strengths and limitations
Strengths:
- Translational design combining human tissue observations with mechanistic mouse and cellular experiments.
- Use of genetic PAD4 deletion to demonstrate causality in vivo.
- Comprehensive phenotyping of cardiac and renal structure and function.
Limitations:
- STZ-induced diabetes predominantly models type 1–like beta cell loss; pathophysiology may differ from type 2 diabetes, which accounts for most human cardiorenal disease.
- Global PAD4 knockout may have developmental or systemic immune effects distinct from pharmacologic inhibition in adults; cell-specific (neutrophil-restricted) approaches would refine attribution.
- Human data were limited to a small EMB cohort (n = 20); larger human studies are necessary to quantify associations across diabetes phenotypes and stages.
- Therapeutic safety concerns: targeting NETosis or PAD4 could impair host defence against infections; clinical translation will require careful risk–benefit assessment.
Future directions
Priority next steps include:
- Validation of NET and inflammasome biomarkers in larger human cohorts with diabetes and prospective correlation with cardiorenal outcomes.
- Preclinical testing of selective PAD4 inhibitors and NET-degrading strategies in models of type 2 diabetes and comorbidities, with assessment of host defence and infection risk.
- Exploration of combination strategies—e.g., PAD4/NET-targeted therapy plus SGLT2 inhibitors or anti‑fibrotic agents—to assess additive or synergistic cardiorenal protection.
- Early-phase clinical trials in carefully phenotyped diabetic populations with endpoints for cardiac and renal structure, function and biomarkers of NETosis/inflammation.
Conclusion
The study by Schommer and colleagues provides compelling experimental evidence that PAD4-dependent NET formation and neutrophil inflammasome activation are mechanistic links between hyperglycaemia and cardiorenal inflammation, fibrosis and dysfunction. These findings shift attention toward neutrophil-driven sterile inflammation as an actionable pathway in diabetic complications and support clinical investigation of PAD4/NET-targeted therapies, with careful attention to translational gaps and safety considerations.
Funding and clinicaltrials.gov
The source article lists funding and disclosure statements; readers should consult the original paper (Schommer et al., Eur Heart J. 2025; DOI: 10.1093/eurheartj/ehaf963) for detailed grant and conflict-of-interest information. No registered clinical trials are reported in the summarized preclinical/observational work.
References
Schommer N, et al. Neutrophil extracellular traps and peptidylarginine deiminase 4-mediated inflammasome activation link diabetes to cardiorenal injury and heart failure. Eur Heart J. 2025 Nov 27:ehaf963. doi: 10.1093/eurheartj/ehaf963. PMID: 41307910.
Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532-1535.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134-147.
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immune responses mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853-1862.
Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature. 2011;470(7336): 6-8 (note: representative inflammasome metabolic inflammation reference).
Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119-1131.
Related articles
– Papayannopoulos V. NETs in sterile tissue damage—review articles detailing NETs and organ injury.
– Li P, et al. PAD4 and NETosis—foundational mechanistic studies.
Author note
This article is written for clinicians and translational investigators to summarise and critically interpret the findings of Schommer et al. (2025) in the context of current knowledge on NETs, PAD4 and inflammasome biology. It is not medical advice.