Highlights
- Identification of a specific somatic hypermutation, K31E, in the immunoglobulin light-chain allele IGLV3-21*02 or *03 as the molecular driver of VITT.
- Discovery of adenoviral core protein VII (pVII) as the inciting antigen that triggers the production of cross-reactive anti-PF4 antibodies.
- Demonstration that back-mutating pathogenic antibodies to their germline sequence abolishes their ability to activate platelets and cause thrombosis.
- Evidence that molecular mimicry between pVII and PF4 explains VITT occurrences following both adenoviral vector vaccination and natural adenovirus infection.
Background: The Enigma of Vaccine-Induced Immune Thrombocytopenia and Thrombosis
Vaccine-induced immune thrombocytopenia and thrombosis (VITT) emerged as a rare but devastating complication following the administration of adenoviral vector-based COVID-19 vaccines, specifically ChAdOx1 nCoV-19 (AstraZeneca) and Ad26.COV2.S (Janssen/Johnson & Johnson). Characterized by high-titer antibodies against platelet factor 4 (PF4), severe thrombocytopenia, and unusual site venous or arterial thrombosis, VITT mirrors the pathophysiology of autoimmune heparin-induced thrombocytopenia (HIT) but occurs in the absence of heparin exposure.
Since its initial description in 2021, the medical community has sought to understand why a viral vector would trigger an autoimmune-like response against a host protein. While it was known that PF4—a positively charged protein released from platelet alpha-granules—was the target, the initial inciting antigen and the genetic predisposition of affected individuals remained elusive. Furthermore, the observation of rare “spontaneous” VITT cases following natural adenovirus infections suggested a broader mechanism of molecular mimicry that transcended vaccination alone.
Study Design: Probing the Immunoglobulin Repertoire
To unravel the immunopathogenesis of VITT, Wang and colleagues conducted a comprehensive study utilizing antibody proteomics and genomic sequencing. The researchers analyzed the amino acid sequences of anti-PF4 antibodies from 21 patients with confirmed VITT and performed gene sequencing of the immunoglobulin light-chain hypervariable regions in a larger cohort of 100 patients.
The methodological framework focused on identifying shared “clonotypes”—specific antibody lineages common across different patients. By comparing the antigen-binding fingerprints of anti-PF4 antibodies with those directed against various adenoviral components, the team sought to find a shared structural origin. The study further employed recombinant antibody technology to test the effects of specific mutations and used both in vitro platelet activation assays and in vivo models to confirm pathogenicity.
Key Results: The K31E Mutation and the pVII Connection
The study revealed a striking genetic commonality among VITT patients. Genomic profiling showed a nearly universal reliance on the immunoglobulin light-chain allele IGLV3-21*02 or *03. Within this specific genetic framework, the researchers identified a critical somatic hypermutation: the substitution of lysine (K) with glutamic acid (E) at position 31 (K31E). This mutation significantly altered the binding characteristics of the antibody.
Identification of the Inciting Antigen
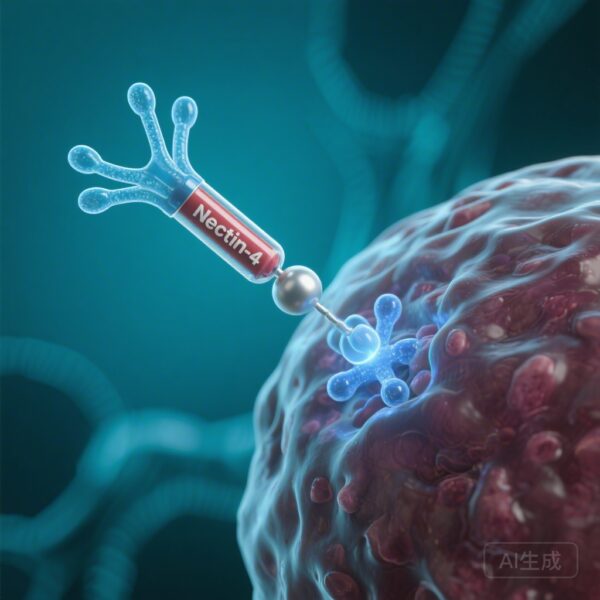
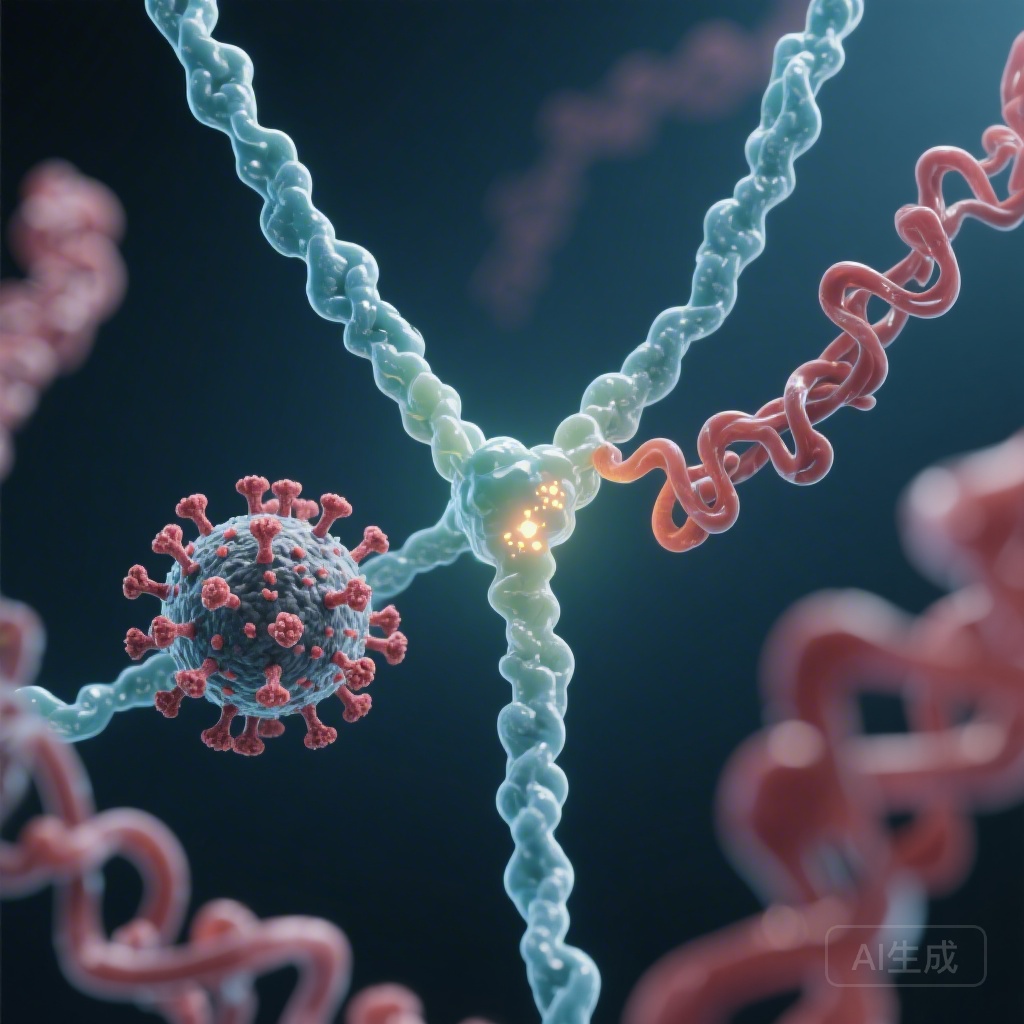
Using proteomic fingerprinting, the researchers tested various adenoviral proteins to see which one shared an epitope with PF4. They found that only antibodies purified against the adenoviral core protein VII (pVII) contained species that matched the VITT anti-PF4 fingerprint. Antibodies against the intact virion or other structural proteins like hexon or penton did not show this cross-reactivity. pVII is a highly basic protein that functions similarly to histones, condensing the viral DNA within the capsid. Its structural and charge characteristics provide the basis for mimicry with the highly cationic PF4.
Functional Validation and Antigenic Shift
To prove the necessity of the K31E mutation, the investigators produced a recombinant version of a pathogenic VITT antibody and then “back-mutated” it to its germline sequence (reverting E31 back to K31). The results were definitive: the germline version lost its ability to bind PF4 and failed to induce platelet activation or thrombosis in experimental models. Interestingly, the germline antibody retained a preference for binding pVII, suggesting that the original immune response was appropriately directed at the virus, but the somatic hypermutation process—intended to refine viral defense—accidentally shifted the antibody’s affinity toward the self-protein PF4.
Mechanistic Insights: From Viral Defense to Autoimmune Catastrophe
The findings support a multi-step model for the development of VITT. First, exposure to adenovirus (either via a vaccine vector or natural infection) introduces the pVII protein to the immune system. In individuals carrying the IGLV3-21*02/03 allele, B cells begin producing antibodies against a linear epitope on pVII. During the process of affinity maturation in the germinal centers, some B cells undergo the K31E mutation.
This single amino acid change—swapping a positive charge for a negative one—creates a paratope that is highly complementary to the positive charge clusters on PF4. Once these cross-reactive antibodies are produced, they bind PF4 to form large immune complexes. these complexes then bind to FcgRIIa receptors on the surface of platelets, leading to massive platelet activation, the release of prothrombotic microparticles, and the clinical manifestation of VITT.
Expert Commentary: Clinical and Research Implications
This study represents a landmark achievement in understanding the intersection of infectious disease and autoimmunity. By identifying a specific genetic marker (the IGLV3-21 allele) and a specific mutation (K31E), it may eventually be possible to identify individuals at higher risk for VITT, although the rarity of the condition makes universal screening impractical at this time.
Refining Vaccine Technology
One of the most significant implications of this research is for future vaccine design. Understanding that pVII is the inciting antigen allows developers to modify adenoviral vectors to reduce the exposure or accessibility of this protein to the immune system. This could potentially preserve the efficacy of adenoviral platforms—which are crucial for global health due to their stability and ease of manufacture—while eliminating the risk of VITT.
Limitations and Generalizability
While the study provides a robust explanation for the majority of VITT cases, it remains to be seen if other minor pathways exist. Additionally, the factors that trigger the specific K31E mutation in some individuals but not others with the same genetic background remain unknown. Environmental factors, the magnitude of the initial viral load, and the specific inflammatory milieu at the time of vaccination likely play secondary roles.
Conclusion: A Definitive Link
The research by Wang et al. closes a major gap in our understanding of VITT. It demonstrates that the condition is not a random side effect but a precise immunological event driven by molecular mimicry. By pinpointing the role of the IGLV3-21 allele and the somatic hypermutation K31E, the study provides a clear biological explanation for why a viral protein can lead to a life-threatening autoimmune response against PF4. This discovery not only enhances our management of VITT but also provides a template for investigating other vaccine-related or infection-triggered autoimmune phenomena.
Funding and Clinical Trial Information
This study was supported by the Deutsche Forschungsgemeinschaft (DFG) and several other international research organizations. The research is registered with the German Clinical Trials Register (DRKS00025738) and the EU Post-Authorization Study Register (EUPAS45098).
References
1. Wang JJ, Schönborn L, Warkentin TE, et al. Adenoviral Inciting Antigen and Somatic Hypermutation in VITT. N Engl J Med. 2026;394(7):669-683. doi:10.1056/NEJMoa2514824.
2. Greinacher A, Thiele T, Warkentin TE, et al. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N Engl J Med. 2021;384(22):2092-2101.
3. Schultz NH, Sørvoll IH, Michelsen AE, et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N Engl J Med. 2021;384(22):2124-2130.