ハイライト
– 糖尿病は、人間とマウスにおいて、心筋内的好中球細胞外トラップ(NET)の負荷を増加させ、好中球インフラマソームの活性化を促進します。
– マウスでのPAD4欠損は、グルコース誘発性NETosisとASC斑点形成を防ぎ、IL-1β、vWF、好中球浸潤、線維症を減少させ、心臓と腎臓の機能を保存します。
– 本研究では、PAD4依存性NET形成とインフラマソームシグナル伝達が、糖尿病性心腎障害のメカニズム的な仲介因子であることを示し、PAD4/NETを潜在的な治療標的として特定しました。
背景:疾患負荷と未満足な需要
心血管疾患と慢性腎臓病は、糖尿病の主要な合併症であり、世界中の罹患率、死亡率、医療費の大部分を占めています。糖尿病性心筋症(DCM)と糖尿病性腎症(DKD)は、持続的な低度の炎症、微小血管機能不全、細胞外基質の再構築、進行性の線維症という共通の病理生理学的特徴を持ち、心不全と進行性の腎機能障害につながります。血糖低下療法や最近の心腎保護薬(例:SGLT2阻害薬)の進歩にもかかわらず、残存リスクが大きく、高血糖から臓器線維症への細胞仲介因子はまだ完全には定義されていません。
研究デザイン
参照文献(Schommer et al., Eur Heart J. 2025)では、ペプチジルアルギニンデイミナーゼ4(PAD4)、好中球細胞外トラップ(NET)、インフラマソームの活性化が、人間と実験的糖尿病関連心腎障害における役割について調査されました。
主要要素:
- 人間成分:糖尿病患者と非糖尿病患者の20人の心不全患者の心筋生検(EMB)でNET負荷を分析しました。
- マウス研究:野生型(WT)とPAD4−/−マウスにストレプトゾトシン(STZ)を投与して実験的糖尿病を誘発しました。経時的評価には、血糖値、体重、心臓機能、運動耐容能、肺水腫が含まれました。
- 細胞アッセイ:ヒトとマウスの好中球を高血糖条件下に曝露してNETosisとインフラマソームの活性化(ASC斑点形成)を評価しました。測定には、共焦点顕微鏡、IL-1βとフォン・ヴィレブランド因子(vWF)のELISA、フローサイトメトリ、組織学的線維症評価(シリアスレッド/ファストグリーン)。腎機能はアルブミノーリアと組織学的に評価されました。
主要な知見
本研究では、高血糖が好中球による炎症と臓器線維症につながる一連の観察結果を報告しています。
1. 糖尿病患者の心筋NET負荷の増加
糖尿病患者の心不全患者の心筋生検では、糖尿病がない患者よりもNET負荷が増加しており、糖尿病性心臓障害におけるNETの臨床的意義を支持しています。
2. 高血糖が好中球インフラマソームを活性化する
ヒト好中球を高血糖条件下に曝露すると、ASC斑点形成が確認され、インフラマソームが活性化することが示されました。これにより、高血糖と先天免疫の活性化とのメカニズム的な関連性が明らかになりました。
3. 糖尿病誘発性NETosisとインフラマソーム活性化にはPAD4が必要
STZ投与後、WTとPAD4−/−マウスの両方が高血糖と体重減少を示しましたが、高血糖損傷は同等でした。しかし、糖尿病後のWT好中球のみがNETosisとASC斑点形成が増加しました。PAD4−/−好中球は、高血糖誘発性NET形成とインフラマソームシグナル伝達に抵抗性であり、PAD4が重要な仲介因子であることが示されました。
4. PAD4欠損が糖尿病関連心機能不全と全身炎症を保護する
糖尿病WTマウスのみが、循環中のIL-1βとvWFが上昇し、心臓機能が低下し、運動耐容能が低下し、肺水腫が認められました。糖尿病PAD4−/−マウスは、同等の高血糖にもかかわらず、これらの悪性現象から保護されていました。これらの知見は、PAD4媒介性NETosisが糖尿病における全身炎症と内皮障害を増幅することを示唆しています。
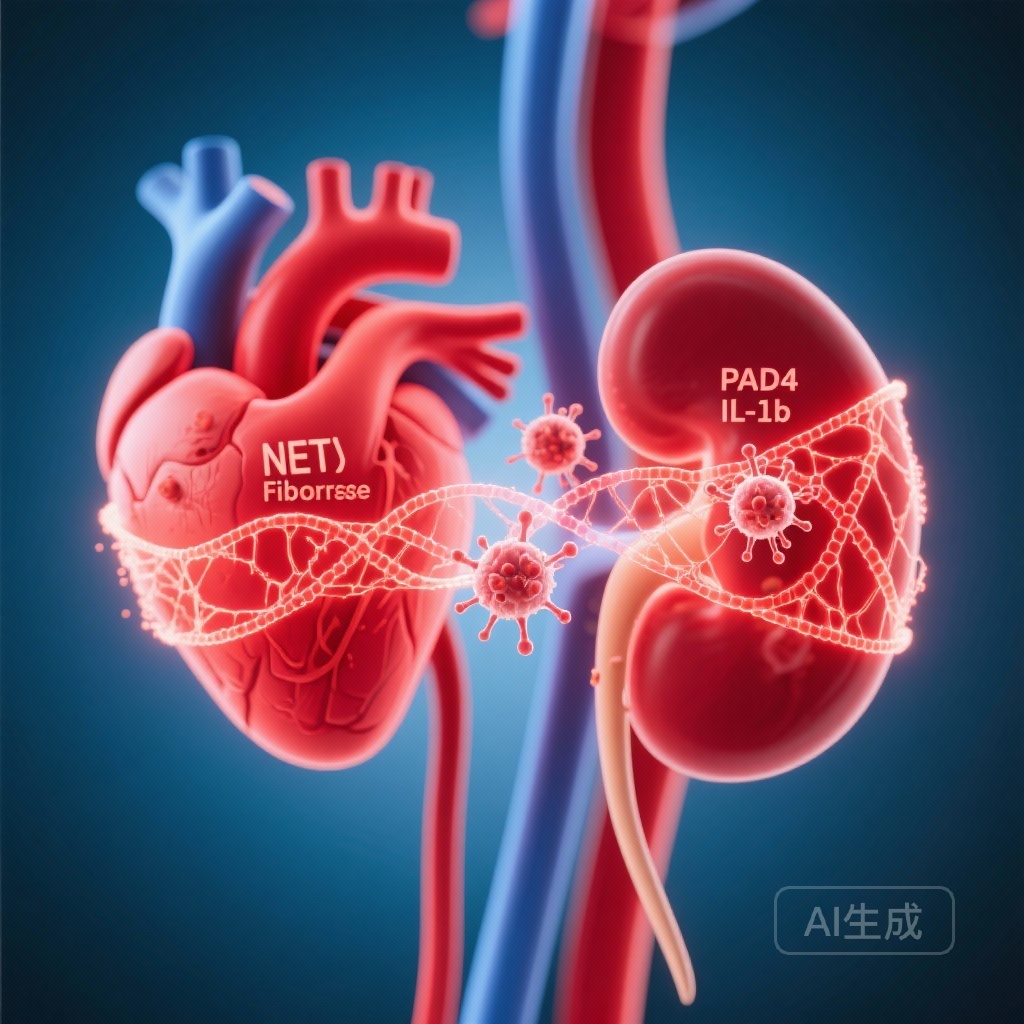
5. PAD4が心臓と腎臓の炎症と線維症を駆動する
組織学的解析では、糖尿病WTマウスでは、より大きな心臓と腎臓の線維症、好中球浸潤、NET沈着、変形成長因子-β1(TGF-β1)レベルの上昇が認められました。機能的には、WTマウスはアルブミノーリアと腎線維症を示すDKDを発症しましたが、糖尿病PAD4−/−マウスは腎機能を保持し、線維症の変化が少なかったことがわかりました。
6. 総合的解釈
総じて、データは、高血糖が好中球インフラマソームの活性化とPAD4依存性NETosisを刺激するモデルを支持しています。NETとインフラマソーム由来のIL-1βは、内皮活性化(vWF放出)を増幅し、より多くの好中球を呼び込み、TGF-β1駆動の線維症再構築を促進し、最終的には心臓と腎臓の機能障害と心不全につながります。
メカニズム的および翻訳的洞察
本研究は、メカニズム的な深さと翻訳的関連性を提供しています。重要なメカニズム的ノードには以下の通りです。
- PAD4媒介性ヒストンシトルリニン化:PAD4はヒストンをシトルリニン化し、クロマチンの脱凝縮とNET放出を促進します。これはNETosisの中心的なステップです。
- 好中球インフラマソーム:ASC斑点形成は、NLRP3などのインフラマソーム複合体の活性化を示し、最終的にはカスパーゼ-1の活性化とIL-1βの放出に至ります。IL-1βは、強力なプロ-線維症性およびプロ-炎症性サイトカインです。
- 血管内皮とプロ線維症性経路との相互作用:NET成分とIL-1βは、内皮障害(vWF放出)とTGF-β1の発現を誘導し、コラーゲン沈着と臓器線維症を促進します。
これらの経路は生物学的に合理的であり、NETが無菌性炎症、血栓症、組織損傷と関連しているという先行文献(Brinkmann et al., 2004; Papayannopoulos, 2018)や、インフラマソーム/IL-1βシグナル伝達が代謝炎症に関与しているという報告(Vandanmagsar et al., 2011)と一致しています。
臨床的および治療的含意
PAD4-NET-インフラマソーム軸の中断から生じる主要な翻訳的機会は以下の通りです。
- PAD4阻害:遺伝的PAD4欠損はマウスで保護効果を示しました。選択的PAD4阻害剤(プレクリニカル/早期臨床開発中)は、糖尿病におけるNETosisと下流の線維症的影響を軽減する可能性があります。
- NET対策戦略:細胞外DNAを分解するDNaseベースの療法やNET形成を阻止する剤は、心腎保護のために再利用または試験される可能性があります。
- インフラマソーム/IL-1β阻害:臨床試験(例:CANTOS, Ridker et al.)は、IL-1β阻害が心血管リスク低下に有効であることを支持しています。本研究は、IL-1β阻害が糖尿病における心腎線維症反応をも制御する可能性があることを示唆していますが、臓器固有の効果は研究が必要です。
- バイオマーカー開発:循環中のNETosis(遊離DNA、シトルリニン化ヒストンH3)とインフラマソーム活性化の指標は、高リスク患者を識別したり、標的療法への反応をモニタリングするためのメカニズム的バイオマーカーとして使用できる可能性があります。
研究の強みと限界
強み:
- 人間組織の観察とメカニズム的マウスおよび細胞実験を組み合わせた翻訳的デザイン。
- in vivoでの因果関係を示すための遺伝的PAD4欠損の使用。
- 心臓と腎臓の構造と機能の包括的な表型化。
限界:
- STZ誘発性糖尿病は主に1型糖尿病様のベータ細胞喪失をモデル化します。病理生理学は、大多数の人間の心腎疾患を占める2型糖尿病とは異なる可能性があります。
- 全身的な免疫効果が成人の薬理学的阻害とは異なる可能性があるため、細胞特異的(好中球限定)アプローチが必要です。
- 人間データは、小さなEMBコホート(n = 20)に限定されており、糖尿病の表型や段階における関連性を量化的に評価するためには、より大規模な人間研究が必要です。
- 治療安全性の懸念:NETosisやPAD4を標的とするのは、感染症に対する宿主防御を阻害する可能性があります。臨床的翻訳には慎重なリスク-ベネフィット評価が必要です。
今後の方向性
優先すべき次のステップには以下の通りです。
- 糖尿病患者の大規模な人間コホートでNETとインフラマソームのバイオマーカーを検証し、心腎アウトカムとの前向き相関を評価します。
- 2型糖尿病と併存症のモデルにおける選択的PAD4阻害剤とNET分解戦略のプレクリニカルテストを行い、宿主防御と感染リスクを評価します。
- 組み合わせ戦略の探索:例えば、PAD4/NET標的療法とSGLT2阻害薬や抗線維症剤の併用で、追加的または相乗的な心腎保護を評価します。
- 慎重に表型化された糖尿病患者を対象とした早期フェーズの臨床試験を行い、心臓と腎臓の構造、機能、NETosis/炎症のバイオマーカーを評価します。
結論
Schommerらの研究は、PAD4依存性NET形成と好中球インフラマソームの活性化が、高血糖と心腎炎症、線維症、機能障害のメカニズム的な関連であるという強力な実験的証拠を提供しています。これらの知見は、糖尿病合併症における好中球による無菌性炎症に注目し、PAD4/NET標的療法の臨床的検討を支持しますが、翻訳的ギャップと安全性に関する注意が必要です。
資金源とclinicaltrials.gov
原著記事には資金源と開示声明が記載されています。詳細な助成金情報や利益相反については、原著論文(Schommer et al., Eur Heart J. 2025; DOI: 10.1093/eurheartj/ehaf963)をご覧ください。要約された前臨床/観察的研究では登録された臨床試験は報告されていません。
参考文献
Schommer N, et al. Neutrophil extracellular traps and peptidylarginine deiminase 4-mediated inflammasome activation link diabetes to cardiorenal injury and heart failure. Eur Heart J. 2025 Nov 27:ehaf963. doi: 10.1093/eurheartj/ehaf963. PMID: 41307910.
Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532-1535.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134-147.
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immune responses mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853-1862.
Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature. 2011;470(7336): 6-8 (注:代表的なインフラマソーム代謝炎症の参考文献).
Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119-1131.
関連記事
– Papayannopoulos V. NETs in sterile tissue damage—無菌性組織損傷におけるNETに関するレビュー記事。
– Li P, et al. PAD4 and NETosis—基礎的なメカニズム研究。
著者注
本記事は、現在のNET、PAD4、インフラマソーム生物学の知識の文脈で、Schommer et al. (2025)の知見を要約し、批判的に解釈するために、臨床医と翻訳研究者向けに書かれています。これは医療アドバイスではありません。