ハイライト
– 稀少なミスセンス変異がCPD(カボキシペプチダーゼD)に存在し、主に感音性の先天性聴覚障害と関連している。
– 致命的な変異はCPDの触媒活性を阻害し、アルギニン/NO/cGMPシグナル伝達を減少させ、聴覚細胞でのERストレス、酸化損傷、およびアポトーシスを促進する。
– モデル生物および細胞モデルにおいて、CPD欠乏による聴覚機能障害はアルギニン補給や薬理学的cGMP増強(シルデナフィル)によって部分的に救済され、治療可能な治療経路を示唆している。
背景と臨床的文脈
先天性聴覚障害(HL)は臨床的にも遺伝的にも多様で、約1,000人に1〜3人が影響を受け、コミュニケーション、発達、生活の質に生涯にわたる影響を及ぼします。遺伝的原因は先天性感音性難聴の大部分を占めており、多くの遺伝子(GJB2、SLC26A4、MYO7Aなど)の病原変異が広範なアレリックおよびメカニズムのスペクトラムを形成しています。遺伝診断が改善されているにもかかわらず、遺伝性HLに対する疾患修飾医療は少なく、管理は主に補助具(補聴器)や手術(人工内耳)およびリハビリテーションに留まっています。新しい遺伝子ベースまたは経路指向の小分子戦略は耳鼻咽喉科における重要な未充足の需要です。
研究デザインと方法(要約)
主要な研究(Ramzan et al., J Clin Invest 2025)では、統合された人間遺伝学と機能生物学的手法を用いて、CPDの機能喪失が先天性HLと関連することを示しました。主な要素には以下のものが含まれます:
- 先天性難聴のある3つの無関係な家族から分離されるミスセンスCPD変異体の同定。
- 10万人ゲノムプロジェクトのコホートを分析し、HLを持つ個体における希少な蛋白質変異型のCPD変異体の富集を検証。
- マウスの耳小骨でCpdの局在化を調査し、感覚上皮および神経成分での表現を地図化。
- 患者由来線維芽細胞でのCPD酵素活性および下流代謝物(アルギニン、一酸化窒素(NO)、cGMP)の生化学的アッセイ。
- Cpd沈黙後の線維芽細胞および組織培養耳小骨での細胞および組織アッセイ(ERストレスマーカー、酸化ストレス、アポトーシス)。
- ジョンストン器官(果蠅の聴覚器官)への影響、行動/運動表現型、アルギニンまたはシルデナフィルを使用した薬理学的救済実験を調査する果蠅モデル。
主要な知見
遺伝子とヒューマンエビデンス
研究者たちは、先天性的に大部分対称的な感音性難聴を持つ3つの無関係な家族の5人の個人から、触媒活性部位CP-domain2にマッピングされる3つの異なるミスセンス変異を発見しました。これらの変異は家族内で疾患と共隔離しており、集団データベースでは稀少でした。10万人ゲノムプロジェクトデータセットの広範な分析では、HLが注釈付けられた参加者の中で希少な蛋白質変異型のCPD変異体の統計的な富集が明らかになり、CPDが本物のHL関連遺伝子であることを支持しました。
局在化と生理学的役割
マウスの耳小骨では、Cpdは感覚上皮細胞(毛細胞および支持細胞に対応する領域を含む)と蝸牛神経節の神経細胞の両方に局在していました。この表現パターンは、聴覚経路での感覚伝達とシナプス/神経の恒常性の両方の役割と一致しています。
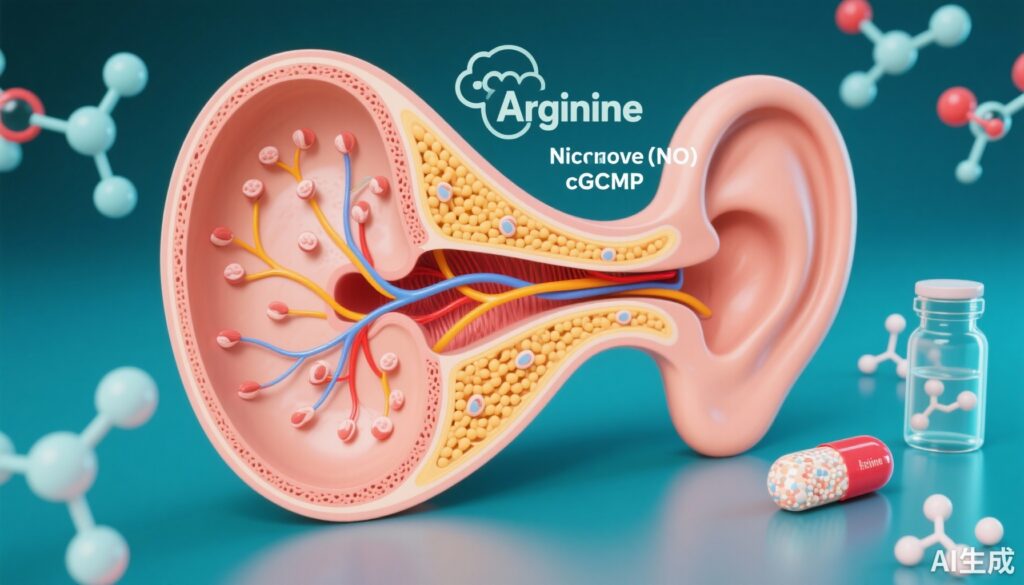
生化学的メカニズム:アルギニン–NO–cGMP軸
CPDはペプチド処理に関与するカボキシペプチダーゼであり、著者らは特定されたCPD変異体が酵素活性を大幅に低下させることを示しました。影響を受けた個人由来の線維芽細胞では、アルギニン、NO、cGMPのレベルが対照群と比較して低下していました。アルギニンから合成されるNOが可溶性グアニル酸シクラーゼを活性化して細胞内cGMPを上昇させるため、NO/cGMPシグナル伝達の低下は、CPD活性の障害と聴覚組織内の細胞機能障害との間の合理的なメカニズム的リンクを提供します。
細胞的影響:ERストレス、酸化ストレス、アポトーシス
患者由来の線維芽細胞では、エンドプレイス網(ER)ストレスのバイオマーカー、酸化ストレスの亢進、細胞死の増加が観察されました。Cpd沈黙後の組織培養マウス耳小骨では、感覚上皮内のアポトーシスが増加しており、同じ細胞死経路が耳小骨組織でも関与していることが示唆されます。これらのデータは、不全なペプチド/アルギニン処理からNOシグナル伝達の障害、レドックスバランスの乱れ、ERストレス介在の聴覚細胞のアポトーシスへの経路を示唆しています。
体内モデリングと薬理学的救済
機能不全のCPDを欠く果蠅モデルでは、ジョンストン器官の構造的欠陥、聴覚伝達の障害、感覚駆動の行動の変化が観察されました。重要的是、アルギニン(NO合成の基質を回復させる)とシルデナフィル(リン酸ジエステラーゼ5阻害剤でcGMPの分解を抑制しcGMPを増加させる)の2種類の介入が、果蠅と一部のex vivoアッセイで部分的に現象を救済しました。これらの介入はすべての測定値を完全に正常化しなかったものの、NO–cGMPシグナル伝達の増強がCPD欠乏関連の聴覚機能障害を改善できるという仮説を支持しています。
解釈と翻訳的意義
この研究は、CPDを先天性感音性聴覚障害の新しい遺伝的原因として確立し、アルギニン依存性NO合成の障害とその下流のcGMPシグナル伝達の失敗により、聴覚組織内の細胞ストレスとアポトーシスが生じるという治療可能なメカニズムを明確にしました。翻訳的な視点からは、NO/cGMPシグナル伝達を高める既存の臨床利用可能な介入(栄養補助のアルギニンと薬理学的cGMP増強剤であるシルデナフィル)を早期フェーズ試験でテストする候補療法として再利用することが最も直ちに行える結果です。無脊椎動物とex vivoモデルでの部分的な救済は重要な概念証明を提供していますが、哺乳類の聴覚システム全体での予臨床効果を証明するには十分ではありません。
専門家のコメント、注意点、制限事項
この研究の強みには、ヒト遺伝学、大規模集団の富集分析、機構的生化学、および種横断的な機能検証の組み合わせが含まれます。CPD下流の定義された代謝-シグナル伝達経路の同定は、生物学的な妥当性を高め、単なる診断上の興味深い遺伝子ではなく、合理的な治療的アプローチを開きます。
重要な制限事項と注意点:
- 患者数は少ないため、表現型スペクトラムはまだ定義されていません。浸透率、進行度、および異なるCPDアレルの重症度の変動については、より大規模なコホート研究と前向きの自然史データが必要です。
- 線維芽細胞はアクセス可能ですが、耳小骨の毛細胞や蝸牛神経節の神経細胞ではありません。周辺細胞株からの推論は内耳の生理学に内在する限界があります。組織培養耳小骨はリンクを強化しますが、完全な哺乳類の体内救済データ(ネズミの遺伝モデル)は重要な次のステップです。
- アルギニン補給とPDE5阻害剤には全身的な影響があります。安全性、最適投与量、タイミング(新生児期、幼児期、それ以降)、および潜在的なオフターゲットの結果は、特に乳児や幼児への使用前に特徴づけなければなりません。
- モデルでの部分的な救済は、治療ウィンドウが狭い可能性があることを示唆しています(細胞死の予防には早期介入が必要かもしれません)し、完全な回復には併用または遺伝子標的戦略が必要である可能性があります。
臨床的および研究的意義 — 提案される進め方
即時優先事項には以下のものがあります:
- 先天性聴覚障害コホートの広範な遺伝子スクリーニングを行い、CPD変異の頻度とアレルスペクトラムを定義する。
- ロッドエントCpdノックアウトまたはノックインモデルの開発を行い、縦断的な表現型評価と予臨床治療試験、特に未熟な哺乳類におけるアルギニンとシルデナフィルの薬物動態と安全性を実施する。
- 生物マーカー(全身または局所のNO/cGMPレベル、ERストレス署名など)の同定と検証を行い、患者選択と薬物動態効果のモニタリングを行う。
- 遺伝学的に確認されたCPD欠損患者を対象とした慎重に段階化された早期フェーズ臨床試験(第1/2相)の設計:短期目標には安全性、耐容性、生物マーカーの調整が含まれ、効果評価項目は客観的な聴力測定(聴覚脳幹反応、耳音響放射)と発達/機能的アウトカムの組み合わせであるべきです。
結論
Ramzan et al. (J Clin Invest 2025) は、CPDを人間の難聴遺伝子のカタログに追加し、重要なことは、アルギニン–NO–cGMPという既存の治療法で標的とすることができるメカニズム経路を明らかにしました。これにより、少なくとも一部の遺伝性聴覚障害のパラダイムは純粋に診断的なものから治療可能なものに変わり、分子診断が直接再利用可能な治療法を示す翻訳的経路を例示しています。ただし、臨床応用までには、大規模な遺伝学的研究、哺乳類の予臨床モデル、安全性プロファイル、効果、タイミング、長期的な結果を確立する慎重に設計された臨床試験などの複数のステップが残っています。
資金源と臨床試験
資金源と詳細な謝辞は元の記事(Ramzan et al., J Clin Invest 2025)に報告されています。出版時点で、アルギニンやPDE5阻害剤をCPD関連の聴覚障害に対して使用した登録臨床試験は知られていません。ヒト試験の前に試験登録が必要です。
参考文献
1. Ramzan M, Ortiz-Vega N, Zafeer MF, Lobato AG, Atik T, Abad C, Vadgama N, Duman D, Bozan N, Avcı Durmuşalioǧlu E, et al. Carboxypeptidase D deficiency causes hearing loss amenable to treatment. J Clin Invest. 2025 Sep 30:e192090. doi: 10.1172/JCI192090. Epub ahead of print. PMID: 41026541.
2. Smith RJH, Bale JF Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–890. doi:10.1016/S0140-6736(05)70853-5.
3. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature Genetics. 1997;15(4): 444–447. doi:10.1038/ng0497-444.
著者注
この記事はRamzan et al.の報告された知見を要約し解釈し、それを臨床的および翻訳的な文脈に位置付けています。医師は前臨床報告に基づいて患者管理を変更すべきではありません。遺伝カウンセリングと多学科的なケアは先天性聴覚障害に影響を受ける家族にとって不可欠です。