Highlights
– Rare missense variants in CPD (carboxypeptidase D) are associated with congenital, predominantly sensorineural hearing loss.
– Pathogenic variants impair CPD catalytic activity, reduce arginine/NO/cGMP signaling, and promote ER stress, oxidative injury and apoptosis in auditory cells.
– In animal and cellular models, auditory dysfunction from CPD deficiency is partially rescued by arginine supplementation or pharmacologic cGMP augmentation (sildenafil), suggesting an actionable therapeutic pathway.
Background and clinical context
Congenital hearing loss (HL) is clinically and genetically heterogeneous, affecting approximately 1–3 per 1,000 newborns and carrying lifelong consequences for communication, development and quality of life. Genetic etiologies account for a large fraction of congenital sensorineural deafness; pathogenic variants in numerous genes (GJB2, SLC26A4, MYO7A, and others) establish a wide allelic and mechanistic spectrum. Despite improved genetic diagnosis, disease-modifying medical therapies for genetic HL are scarce, and most management remains prosthetic (hearing aids) or surgical (cochlear implantation) and rehabilitative. New gene-based or pathway-directed small-molecule strategies are an important unmet need in otology.
Study design and methods (summary)
The primary study (Ramzan et al., J Clin Invest 2025) used an integrated human-genetics and functional biology approach to link CPD loss-of-function to congenital HL. Key elements included:
- Identification of segregating missense CPD variants in three unrelated families with congenital deafness.
- Analysis of a larger cohort (100,000 Genomes Project) to test for enrichment of rare, protein-altering CPD variants among individuals with HL.
- Localization studies in mouse cochlea to map Cpd expression in sensory epithelium and neuronal elements.
- Biochemical assays of CPD enzymatic activity and downstream metabolites (arginine, nitric oxide (NO), cGMP) in patient-derived fibroblasts.
- Cellular and tissue assays (ER stress markers, oxidative stress, apoptosis) in fibroblasts and organotypic cochlear cultures after Cpd silencing.
- Drosophila models to probe effects on the Johnston’s organ (the fly auditory organ), behavioral/locomotor phenotypes and pharmacologic rescue experiments with arginine or sildenafil.
Key findings
Genetics and human evidence
Investigators discovered three distinct missense variants mapping to the catalytically active CP-domain2 of CPD in five individuals from three unrelated families with congenital, largely symmetric sensorineural deafness. These variants co-segregated with disease in the families and were rare in population databases. Broader analysis of the 100,000 Genomes Project dataset revealed a statistical enrichment of rare protein-altering CPD variants among participants annotated with HL, supporting CPD as a bona fide HL-associated gene.
Localization and physiological role
In mouse cochlea, Cpd localized to both sensory epithelial cells (including regions corresponding to hair cells and supporting cells) and to neurons in the spiral ganglion. This expression pattern is consistent with a role in both sensory transduction and synaptic/neuronal homeostasis in the auditory pathway.
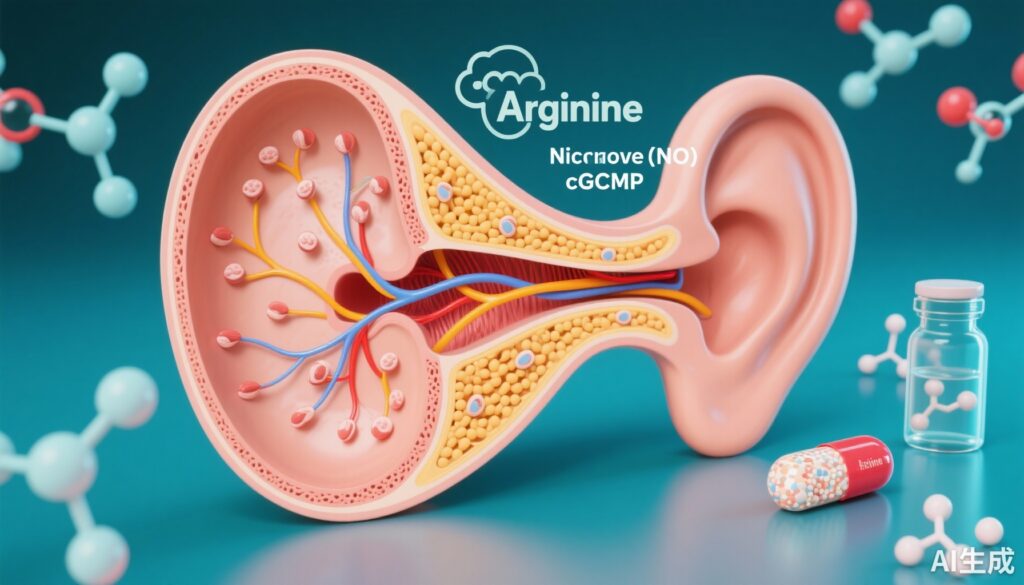
Biochemical mechanism: arginine–NO–cGMP axis
CPD is a carboxypeptidase implicated in peptide processing; the authors showed that the identified CPD variants appreciably reduced enzymatic activity. In fibroblasts from affected individuals, levels of arginine, NO and cGMP were decreased compared with controls. Since NO synthesized from arginine activates soluble guanylate cyclase to raise intracellular cGMP, reduction in NO/cGMP signaling provides a plausible mechanistic link between impaired CPD activity and cellular dysfunction within auditory tissues.
Cellular consequences: ER stress, oxidative stress, apoptosis
Patient fibroblasts demonstrated biomarkers of endoplasmic reticulum (ER) stress, elevated oxidative stress and increased cell death. In organotypic mouse cochlea cultures, silencing Cpd produced increased apoptosis within the sensory epithelium, implicating the same cell-death pathways in cochlear tissue. Collectively, these data suggest a path from impaired peptide/arginine processing to disrupted NO signaling, redox imbalance and ER stress–mediated apoptosis of auditory cells.
In vivo modeling and pharmacologic rescue
Drosophila models lacking functional CPD showed structural defects in Johnston’s organ, impaired auditory transduction, and altered sensory-driven behaviors. Importantly, two types of interventions partially rescued phenotypes in flies and in some ex vivo assays: supplementation with arginine (to restore substrate for NO synthesis) and administration of sildenafil (a phosphodiesterase type 5 inhibitor that increases cGMP by reducing its breakdown). These interventions did not fully normalize all measures, but the partial rescue supports the hypothesis that augmenting NO–cGMP signaling can ameliorate CPD-deficiency–related auditory dysfunction.
Interpretation and translational implications
This work establishes CPD as a novel genetic cause of congenital sensorineural hearing loss and clarifies a tractable mechanism — impaired arginine-dependent NO synthesis with downstream failure of cGMP signaling, leading to cellular stress and apoptosis in auditory tissues. From a translational perspective, the most immediately actionable consequence is that existing, clinically available interventions that elevate NO/cGMP signaling (nutritional arginine supplementation and pharmacologic cGMP enhancers such as sildenafil) could be repurposed as candidate therapies to test in early-phase trials. The partial rescue observed in invertebrate and ex vivo models provides important proof-of-concept but falls short of conclusive preclinical efficacy across mammalian auditory systems.
Expert commentary, caveats and limitations
Strengths of the study include the combination of human genetics, population- scale enrichment analysis, mechanistic biochemistry and cross-species functional validation. The identification of a defined metabolic-signaling pathway downstream of CPD enhances biological plausibility and opens a rational therapeutic avenue rather than leaving the gene as a diagnostic curiosity.
Key limitations and points of caution:
- Patient numbers are small and the phenotypic spectrum remains to be defined. Penetrance, progression, and variability in severity for different CPD alleles require larger cohort studies and prospective natural-history data.
- Fibroblasts are accessible but are not cochlear hair cells or spiral ganglion neurons; extrapolation from peripheral cell lines to inner-ear physiology has inherent limits. While organotypic cochlea cultures strengthen the link, full mammalian in vivo rescue data (rodent genetic models) would be an important next step.
- Arginine supplementation and PDE5 inhibitors have systemic effects. Safety, optimal dosing, timing (neonatal vs early childhood vs later), and potential off-target consequences must be characterized before clinical use, particularly in infants and young children.
- Partial rescue in models suggests that treatment windows may be narrow (prevention of cell death might require early intervention) and that combination or gene-targeted strategies may be necessary for complete recovery.
Clinical and research implications — a proposed path forward
Immediate priorities include:
- Broader genetic screening of congenital HL cohorts to define prevalence and allele spectrum of CPD variants.
- Development of rodent Cpd knockout or knock-in models for longitudinal phenotyping and preclinical therapeutic testing, including pharmacokinetics and safety of arginine and sildenafil in immature mammals.
- Identification and validation of biomarkers (e.g., systemic or local NO/cGMP levels, ER-stress signatures) to select patients and monitor pharmacodynamic effects.
- Design of a carefully staged early-phase clinical trial (Phase 1/2) in genetically confirmed CPD-deficient patients: short-term objectives could include safety, tolerability and biomarker modulation; efficacy endpoints should combine objective audiometric measures (auditory brainstem responses, otoacoustic emissions) with developmental/functional outcomes.
Conclusion
Ramzan et al. (J Clin Invest 2025) add CPD to the catalog of human deafness genes and, critically, reveal a mechanistic pathway — arginine–NO–cGMP — that is targetable with existing therapies. This alters the paradigm for at least a subset of genetic hearing loss from purely diagnostic to potentially treatable, and exemplifies a translational trajectory where molecular diagnosis can directly inform repurposed therapeutics. Nonetheless, multiple steps remain before clinical application: larger genetic studies, mammalian preclinical models, safety profiling, and carefully designed clinical trials to establish efficacy, timing and long-term outcomes.
Funding and clinical trials
Funding sources and detailed acknowledgements are reported in the original article (Ramzan et al., J Clin Invest 2025). No registered clinical trials of arginine or PDE5 inhibitors specifically for CPD-related hearing loss are known at the time of publication; trial registration would be required prior to human testing.
References
1. Ramzan M, Ortiz-Vega N, Zafeer MF, Lobato AG, Atik T, Abad C, Vadgama N, Duman D, Bozan N, Avcı Durmuşalioǧlu E, et al. Carboxypeptidase D deficiency causes hearing loss amenable to treatment. J Clin Invest. 2025 Sep 30:e192090. doi: 10.1172/JCI192090. Epub ahead of print. PMID: 41026541.
2. Smith RJH, Bale JF Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–890. doi:10.1016/S0140-6736(05)70853-5.
3. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature Genetics. 1997;15(4): 444–447. doi:10.1038/ng0497-444.
Author note
This article summarizes and interprets findings reported by Ramzan et al. and places them in a clinical and translational context. Clinicians should not change patient management based solely on preclinical reports; genetic counseling and multidisciplinary care remain essential for families affected by congenital hearing loss.