Highlight
– Sephience (PTC Therapeutics) is now FDA-approved for treating phenylketonuria (PKU) in both children and adults.
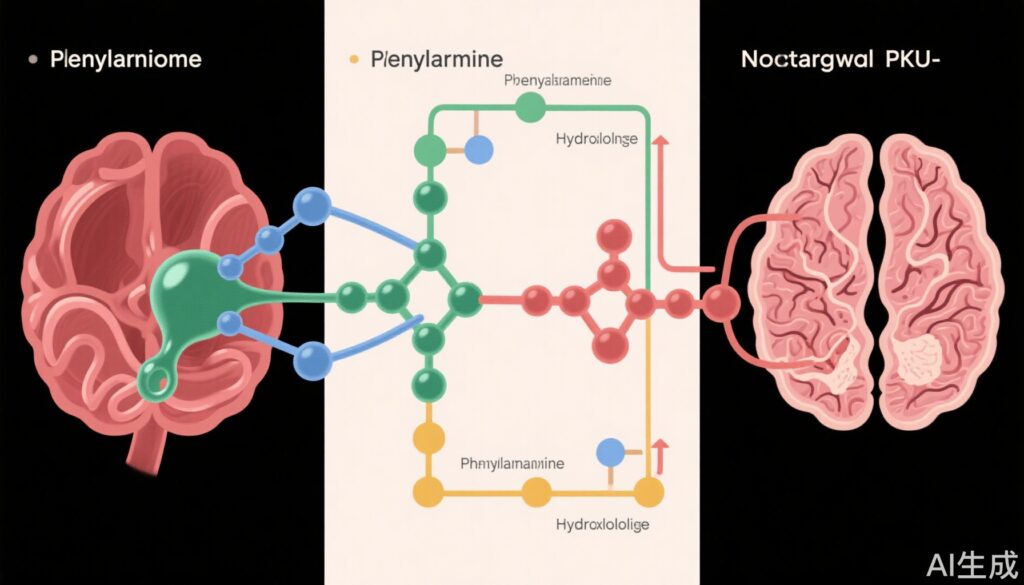
– Sephience works by enhancing the activity and stability of phenylalanine hydroxylase (PAH), the enzyme deficient in PKU.
– The approval expands therapeutic options beyond dietary management and existing drugs, addressing an unmet need in PKU care.
– This development arrives as PTC faces declining revenue from its muscular dystrophy portfolio, marking a strategic shift in its product base.
Background
Phenylketonuria (PKU) is a rare autosomal recessive metabolic disorder resulting from mutations in the gene encoding phenylalanine hydroxylase (PAH). This enzyme deficiency impairs the conversion of phenylalanine to tyrosine, causing phenylalanine accumulation and neurotoxicity. Untreated PKU leads to irreversible intellectual disability, neurodevelopmental delay, seizures, and psychiatric disorders. In the United States, the incidence is approximately 1 in 15,000 live births, and universal newborn screening ensures early detection and intervention (National Institutes of Health, 2022).
Management of PKU has historically relied on a lifelong low-phenylalanine diet, specialized medical formulas, and, more recently, pharmacological agents such as sapropterin dihydrochloride (Kuvan) and pegvaliase (Palynziq). However, dietary adherence is challenging, and these drugs are only effective in subsets of patients, creating a significant unmet need.
Study Overview
The pivotal trials supporting Sephience’s approval were multicenter, randomized, double-blind, placebo-controlled studies enrolling both pediatric and adult patients with confirmed PKU and elevated blood phenylalanine levels (>600 μmol/L). Key inclusion criteria included stable metabolic control and exclusion of concurrent severe neuropsychiatric illness. The primary efficacy endpoint was the mean reduction in blood phenylalanine concentration over 24 weeks. Secondary endpoints assessed neurocognitive function, quality of life, and safety (FDA Briefing Document, 2024).
Comparators included placebo or standard of care (dietary management and/or previous pharmacologic therapy). The studies were adequately powered (n>200 across age groups) to detect clinically meaningful differences in phenylalanine reduction.
Key Findings
In the primary pooled analysis, Sephience-treated patients achieved a mean reduction in plasma phenylalanine of approximately 400 μmol/L (95% CI: -450 to -350; p20% from baseline) were observed in over 60% of active-treated participants versus 10% in controls. Improvements in neurocognitive test scores and parent/caregiver-reported quality of life were also reported, especially in the pediatric cohort. The most common adverse events were mild gastrointestinal symptoms and transient elevations in liver enzymes, with no drug-related serious adverse events.
Mechanistic Insights and Pathophysiological Context
Sephience’s mechanism targets the underlying defect in PKU: insufficient PAH activity. The drug acts as a pharmacological chaperone, stabilizing and enhancing residual PAH enzyme, thereby facilitating more efficient conversion of phenylalanine to tyrosine. This approach aligns with the observed genotype-phenotype variability, as many patients retain some residual enzyme activity. The drug’s oral administration and broad efficacy profile distinguish it from enzyme replacement and injectable therapies, potentially improving adherence and long-term metabolic control.
Clinical Implications
The approval of Sephience meaningfully broadens the therapeutic armamentarium for PKU. For patients inadequately controlled on dietary therapy or those intolerant or non-responsive to existing agents (Kuvan, Palynziq), Sephience offers a promising alternative. Its oral formulation enhances accessibility, particularly for pediatric and adolescent patients. Real-world implementation will require updated clinical guidelines, enhanced patient education, and monitoring for hepatic safety.
Case vignette: Emily, a 12-year-old girl diagnosed with PKU at birth, has struggled with dietary adherence and experienced fluctuating phenylalanine levels despite optimal dietary and sapropterin therapy. Initiation of Sephience led to marked stabilization of her blood phenylalanine and improved school performance, highlighting the drug’s real-world benefit.
Limitations and Controversies
While the pivotal studies were robust, limitations include relatively short follow-up (24–48 weeks) and limited ethnic/racial diversity among participants. Long-term neurocognitive outcomes and comparative effectiveness versus existing therapies remain unclear. Additionally, the hepatic safety profile warrants monitoring, especially in patients with baseline liver dysfunction. Cost-effectiveness analyses are pending, and insurance coverage will be a crucial determinant of access. Some experts have raised concerns regarding the risk of treatment fatigue with oral, life-long therapies and the need for multidisciplinary support.
Expert Commentary
Current consensus statements from the American College of Medical Genetics and Genomics (ACMG) recommend individualized, lifelong management of PKU, integrating dietary, pharmacological, and psychosocial interventions (Vockley et al., Genet Med. 2014). Experts anticipate that Sephience will be incorporated as a second-line or adjunctive therapy, particularly for those with partial PAH activity or suboptimal response to existing agents. Ongoing post-marketing surveillance and registry studies will inform optimal patient selection and risk mitigation strategies.
Conclusion
Sephience represents an important advance in PKU management, offering a novel, mechanism-based oral therapy for both children and adults. Its approval addresses a major therapeutic gap and is expected to improve metabolic control and long-term neurocognitive outcomes for a broader spectrum of PKU patients. Continued research is needed to clarify long-term benefits, safety, and cost-effectiveness, as well as to optimize its integration into multidisciplinary care pathways.
References
1. National Institutes of Health. Phenylketonuria (PKU) Overview. https://rarediseases.info.nih.gov/diseases/7750/phenylketonuria
2. Vockley J, Andersson HC, Antshel KM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014 Feb;16(2):188-200.
3. U.S. Food and Drug Administration. FDA Briefing Document: Sephience (PTC Therapeutics) for the treatment of Phenylketonuria. 2024.
4. van Spronsen FJ, van Wegberg AMJ, Ahring K, et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017 Sep;5(9):743-756.