Nhấn mạnh
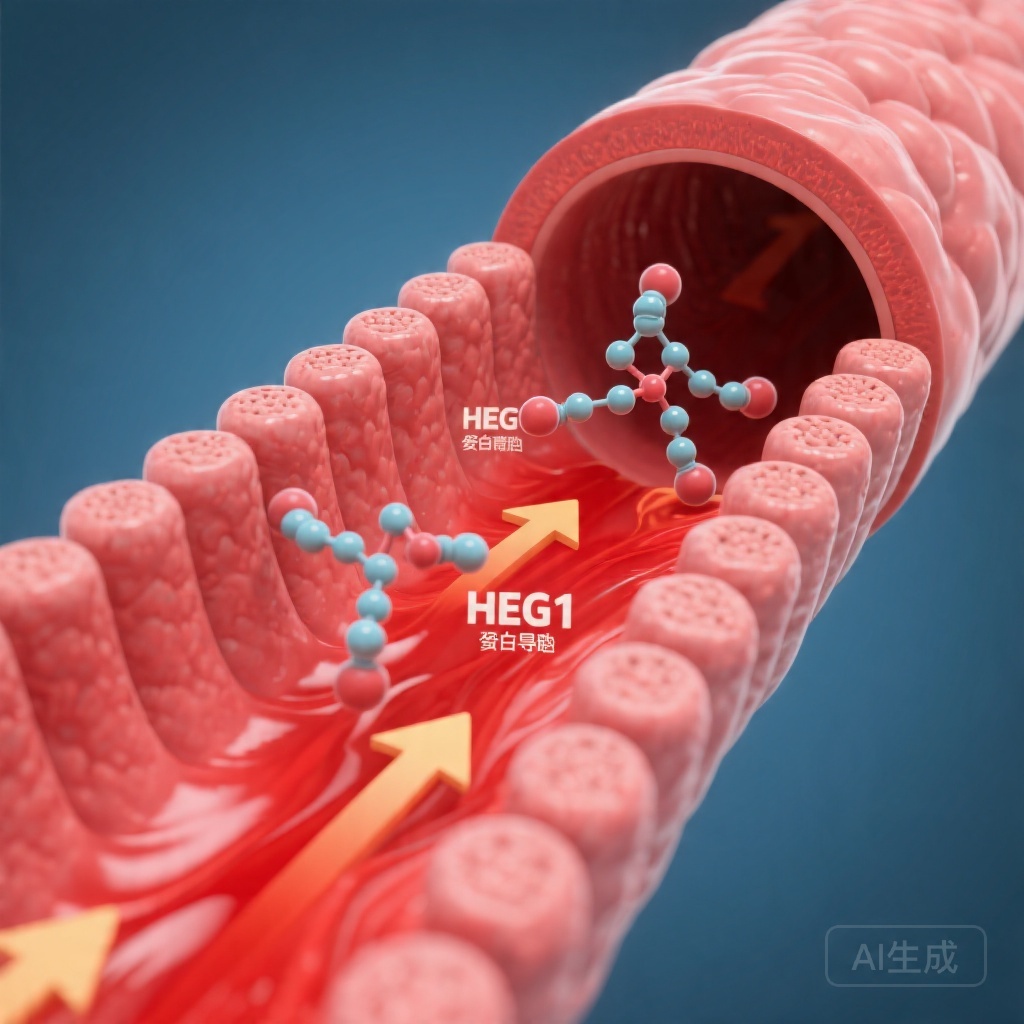
Căng thẳng cắt gây ra tín hiệu nội mô HEG1 đại diện cho một con đường điều hòa mới liên kết động lực học lưu lượng máu với kiểm soát huyết áp toàn thân. Mức độ HEG1 trong huyết tương giảm đáng kể ở bệnh nhân tăng huyết áp do giảm căng thẳng cắt trên nội mô mạch máu. Xóa Heg1 đặc hiệu nội mô ở chuột dẫn đến tăng huyết áp, suy giảm giãn mạch phụ thuộc nội mô và rối loạn sản xuất NO. Cơ chế này liên quan đến việc HEG1 điều chỉnh sự phân giải PHACTR1, điều khiển quá trình phiên mã eNOS phụ thuộc SP1.
Nền tảng
Tăng huyết áp vẫn là yếu tố nguy cơ có thể thay đổi hàng đầu đối với bệnh tim mạch, đột quỵ và rối loạn chức năng thận trên toàn thế giới. Mặc dù có nhiều loại thuốc chống tăng huyết áp, một tỷ lệ đáng kể bệnh nhân không đạt được kiểm soát huyết áp đầy đủ, nhấn mạnh nhu cầu về mục tiêu điều trị mới. Các tế bào nội mô bao phủ toàn bộ hệ thống mạch máu và hoạt động như các cảm biến cơ học quan trọng, chuyển đổi các lực thủy động, đặc biệt là căng thẳng cắt, thành các tín hiệu sinh hóa điều khiển trương lực mạch và cân bằng huyết áp.
Căng thẳng cắt, lực ma sát do lưu lượng máu tác động lên bề mặt nội mô, kích thích giãn mạch thông qua các cơ chế chưa được hiểu rõ hoàn toàn. Sự suy giảm của con đường chuyển hóa cơ học này góp phần vào rối loạn chức năng nội mô, một dấu hiệu đặc trưng của tăng huyết áp và bệnh tim mạch xơ vữa. Hiểu biết về các trung gian phân tử kết nối cảm biến căng thẳng cắt với sản xuất nitric oxide (NO) có thể tiết lộ các mục tiêu hành động để quản lý huyết áp.
Protein phát triển tim có miền EGF-like 1 (HEG1) là một protein màng nguồn gốc nội mô trước đây đã được liên kết với động lực học dòng chảy và các biểu hiện tim mạch. Dữ liệu dịch tễ học gợi ý mối quan hệ nghịch đảo giữa biểu hiện HEG1 và các yếu tố nguy cơ tim mạch, nhưng cơ sở cơ chế của mối liên hệ này chưa được xác định. Nghiên cứu hiện tại nhằm làm rõ vai trò của HEG1 nội mô trong điều chỉnh huyết áp và đặc trưng hóa các chuỗi tín hiệu tiềm ẩn.
Thiết kế nghiên cứu
Đề tài sử dụng phương pháp tiếp cận đa chế độ tích hợp dịch tễ học con người, mô hình hóa tính toán và di truyền học chuột thí nghiệm để thiết lập mối quan hệ nhân quả giữa tín hiệu HEG1 và kiểm soát huyết áp.
Các nghiên cứu trên người bao gồm phân tích liên kết phạm vi biểu tượng trên nhiều nhóm, mô hình hóa động lực học chất lỏng tính toán để đặc trưng hóa các mẫu căng thẳng cắt trong động mạch dẫn, phân tích RNA đơn tế bào của tế bào nội mô từ mẫu động mạch và đo nồng độ HEG1 trong huyết tương ở bệnh nhân tăng huyết áp và huyết áp bình thường từ các nhóm xác minh độc lập.
Các nghiên cứu thí nghiệm sử dụng chuột gõ mất Heg1 đặc hiệu nội mô được tạo ra thông qua tái tổ hợp Cre-lox dưới sự điều khiển của启动子Tie2。使用遥测技术在清醒、不受限制的动物中进行血压监测。通过线性张力法评估血管功能,以评估内皮依赖性和内皮非依赖性扩张。研究在C57BL/6J和载脂蛋白E敲除(ApoeKO)背景中进行,以检查与血脂异常的相互作用。
机制研究采用蛋白质组学、转录组学、泛素化分析和核质分离来识别HEG1相互作用蛋白和下游效应因子。使用CCG-1423抑制PHACTR1核转运测试了治疗潜力。
主要发现
人类高血压中的HEG1减少
在三个独立队列中,高血压患者的血浆HEG1浓度显著低于正常血压对照组(p < 0.001)。这种减少与通过计算流体动力学分析颈动脉和肱动脉得出的壁面剪切应力估计值降低相关。单细胞RNA测序证实HEG1表达主要在内皮细胞中,在低剪切应力区域暴露的血管中转录水平降低。
Heg1删除后的血压升高
内皮特异性Heg1敲除小鼠的收缩压持续升高,与同窝对照组相比(平均差异:+12 mmHg,95% CI:8-16 mmHg,p < 0.001)。在ApoeKO背景下,这种血压升高尤为明显,Heg1缺陷小鼠发展出相当于人类2级高血压的高血压。舒张压和平均动脉压同样升高。
扩张能力受损
线性张力法显示,Heg1缺陷主动脉和肠系膜阻力动脉的内皮依赖性扩张严重受损。乙酰胆碱剂量反应曲线向右移动,最大反应减少约60%(p < 0.001)。内皮非依赖性对硝普钠的扩张保持完整,证实了一种特定的内皮机制。在C57BL/6J小鼠中基线时观察到损害,并在高脂饮食下在ApoeKO动物中加剧。
分子机制:HEG1-PHACTR1-eNOS轴
蛋白质组学分析确定PHACTR1(磷酸酶和肌动蛋白调节蛋白1)是一种结合伙伴,其水平受HEG1的相反调节。HEG1与CUL3物理相互作用,CUL3是Cullin-RING E3泛素连接酶复合物的支架蛋白,促进CUL3介导的PHACTR1泛素化和蛋白酶体降解。在缺乏HEG1的情况下,PHACTR1积累并发生核转运。
在细胞核内,PHACTR1抑制SP1介导的eNOS(内皮一氧化氮合酶)转录,后者是一氧化氮产生的限速酶。染色质免疫沉淀证实Heg1缺陷内皮细胞中eNOS启动子上的SP1结合减少。相应地,条件培养基和主动脉组织中的一氧化氮代谢物水平显著降低。
药物挽救
用CCG-1423处理,一种小分子抑制剂,可防止Heg1缺陷小鼠的血压升高并恢复内皮依赖性扩张。通过饮用水给药四周减少核PHACTR1积累,将eNOS表达恢复到野生型水平,并使血压恢复正常,而不影响收缩功能或肾参数。
关键机制途径
正常剪切应力 → HEG1表达和释放 → CUL3介导的PHACTR1降解 → 核内PHACTR1低水平 → 不受抑制的SP1活性 → eNOS转录 → NO产生 → 扩张 → 正常血压。
高血压 ↓ 剪切应力 → HEG1下调 → PHACTR1积累 → 核转运 → SP1抑制 → eNOS抑制 → NO减少 → 扩张受损 → 血压升高。
专家评论
这些发现确立了HEG1作为关键的机械敏感调节因子,将水动力力与内皮NO产生联系起来。HEG1介导的泛素化控制剪切应力和PHACTR1核定位之间的逆关系,为低或振荡剪切应力区域如何导致内皮功能障碍和高血压提供了令人信服的分子解释。
观察到药物抑制PHACTR1核输入在遗传性高血压模型中恢复正常血压表明该途径可能适合治疗靶向。尽管CCG-1423目前尚未获得临床批准,但它代表了一类新型抗高血压药物的概念验证。
几个局限性值得考虑。首先,人类研究具有相关性;虽然遗传小鼠模型支持其转化意义,但需要进行人类干预研究。其次,ApoeKO背景虽然与动脉粥样硬化疾病相关,但引入了代谢混杂因素。第三,鉴于PHACTR1在其他组织(包括大脑和肾脏)中的作用,长期安全性的PHACTR1抑制仍有待确定。
HEG1作为支架将CUL3和PHACTR1聚集在一起的机制见解扩展了我们对内皮机械传导的理解。是否基于HEG1的疗法可以补充现有的抗高血压策略,特别是在耐药性高血压患者中,值得研究。
结论
本研究确定了一个新的剪切应力响应信号轴,中心在于内皮HEG1,通过调节PHACTR1泛素化、核定位和下游eNOS表达来控制血压。在高血压患者中观察到的血浆HEG1减少反映了内皮机械传感的减弱,可能作为受损流量介导扩张的生物标志物。
从剪切应力到HEG1、CUL3、PHACTR1、SP1再到eNOS转录的机制级联代表了一个先前未被认识的途径,将血管生物学与全身血压控制联系起来。CCG-1423介导的PHACTR1核输入抑制为与内皮功能障碍相关的高血压提供了一种潜在的治疗策略。
未来方向包括开发HEG1模拟肽或小分子激活剂,验证血浆HEG1作为预后生物标志物,并在记录有内皮功能障碍的高血压人群中进行PHACTR1核输入抑制剂的临床试验。
资金来源
本研究得到了国家自然科学基金、中国医学科学院和欧洲研究委员会的资助。作者声明没有利益冲突。
参考文献
Wu W, Liu J, Chen X, Zhu P, Xu J, Yue J, Liu X, Fang J, Chen X, Pi J, Zheng L, Zhang Q, Zhang L, Schneider CV, Schneider KM, Trautwein C, Gao P, Reilly MP, Zhang Y, Zheng X, Liu J. 剪切应力诱导的内皮HEG1信号调节血管张力和血压。Eur Heart J. 2026;47(14):1721-1737. PMID: 40986512。