Introduction
Dilated cardiomyopathy (DCM) is a significant cause of heart failure characterized by dilation and impaired contraction of the left or both ventricles. Genetic causes, particularly monogenic variants, contribute to approximately one-third of DCM cases. Clinically, DCM affects males about twice as often as females, but it remains unclear whether this reflects diagnostic bias or genuine differences in genetic susceptibility and disease penetrance between sexes. This study systematically explored sex differences in genetic architecture, focusing on whether specific gene variants exhibit sex-specific penetrance impacts in DCM patients.
Study Design and Methods
The study recruited 902 adult DCM patients prospectively from 11 UK centers, with 830 unrelated patients undergoing whole genome sequencing (WGS) after stringent quality control—of these, 33% were female. Patients met contemporary ESC guidelines for DCM diagnosis, excluding cases explained solely by loading conditions or coronary artery disease. Variant analysis focused on 12 definitive DCM-associated genes, with rare variants (minor allele frequency <0.01%) classified by consequence: predicted loss-of-function (pLoF), missense, and combined protein-altering variants (PAVs). Burden testing compared variant frequencies between males and females, using Fisher’s exact tests and false discovery rate adjustments to identify statistically significant differences.
Furthermore, sex-stratified association analyses of desmoplakin (DSP) gene variants were validated using whole exome sequencing data from 469,397 UK Biobank participants. Penetrance estimates were generated from population allele frequencies in cases versus controls, stratified by sex.
Key Findings
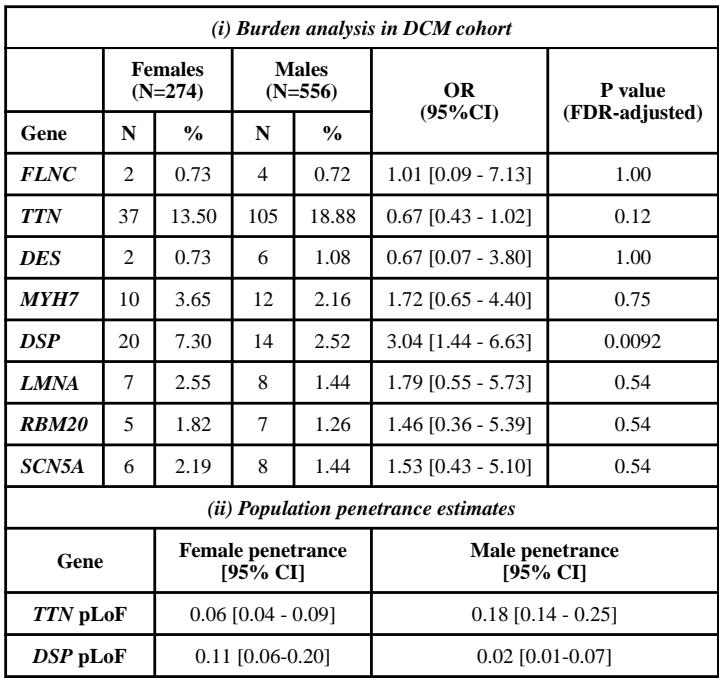
The key finding is that variants in the desmoplakin gene (DSP) are significantly more prevalent and confer a greater risk for DCM in females than males. Specifically, 7.3% of female patients carried DSP protein-altering variants compared to 2.5% of males (OR 3.04, 95% CI 1.44–6.63, P_FDR=0.0092). Similarly, predicted loss-of-function (pLoF) variants in DSP were found in 2.2% of females versus only 0.36% of males (OR 6.19, 95% CI 1.1–63). This female predominance of DSP variants was validated within the UK Biobank cohort, where DSP pLoF variants were tenfold more frequent in female DCM cases compared to males.
| (i) Burden analysis in DCM cohort | ||||||
|---|---|---|---|---|---|---|
| Females (N=274) |
Males (N=556) |
OR (95%CI) |
P value (FDR-adjusted) |
|||
| Gene | N | % | N | % | ||
| FLNC | 2 | 0.73 | 4 | 0.72 | 1.01 [0.09 – 7.13] | 1.00 |
| TTN | 37 | 13.50 | 105 | 18.88 | 0.67 [0.43 – 1.02] | 0.12 |
| DES | 2 | 0.73 | 6 | 1.08 | 0.67 [0.07 – 3.80] | 1.00 |
| MYH7 | 10 | 3.65 | 12 | 2.16 | 1.72 [0.65 – 4.40] | 0.75 |
| DSP | 20 | 7.30 | 14 | 2.52 | 3.04 [1.44 – 6.63] | 0.0092 |
| LMNA | 7 | 2.55 | 8 | 1.44 | 1.79 [0.55 – 5.73] | 0.54 |
| RBM20 | 5 | 1.82 | 7 | 1.26 | 1.46 [0.36 – 5.39] | 0.54 |
| SCN5A | 6 | 2.19 | 8 | 1.44 | 1.53 [0.43 – 5.10] | 0.54 |
| (ii) Population penetrance estimates | ||||||
| Gene | Female penetrance [95% CI] |
Male penetrance [95% CI] |
||||
| TTN pLoF | 0.06 [0.04 – 0.09] | 0.18 [0.14 – 0.25] | ||||
| DSP pLoF | 0.11 [0.06-0.20] | 0.02 [0.01-0.07] | ||||
By contrast, no significant sex differences in variant frequencies were observed for other well-established DCM genes such as LMNA, RBM20, SCN5A, TTN, FLNC, DES, or MYH7. Notably, titin truncating variants (TTNtvs) demonstrated higher penetrance in males (18% vs. 6% female penetrance), reinforcing established male susceptibility in TTN-related DCM.
Penetrance estimation showed DSP pLoF variants have an overall penetrance of about 8%, with a notably higher penetrance in females (11%) than males (2%). These findings imply sex-specific genetic risk profiles, with DSP variants disproportionately affecting female patients with significant clinical ramifications.
Expert Commentary
The identification of sex-specific penetrance in DSP variants is a critical advance illuminating the genetic underpinnings of DCM. While most DCM genes are autosomal, the differential impact on females versus males likely involves complex interactions between genetics, hormonal milieu, and environmental factors. DSP-related cardiomyopathy is associated with adverse outcomes, and female sex has emerged as a marker of poor prognosis within this subgroup, underscoring the need for sex-tailored risk stratification and management approaches.
Male predominance in overall DCM incidence may relate to sex-dependent penetrance of other genes like TTN and non-genetic modulators that warrant further investigation. The study’s limited ancestral diversity restricts broader applicability, suggesting the necessity for diverse cohorts to fully elucidate sex-by-ancestry interactions in DCM genetics.
Understanding the mechanisms driving increased female susceptibility to DSP variant-induced DCM could inform personalized surveillance and therapeutic strategies. Furthermore, reproductive history and hormonal influences represent important unresolved factors that future research should address.
Conclusion
This comprehensive genetic analysis reveals that desmoplakin gene variants confer a significantly higher risk and penetrance of dilated cardiomyopathy in females compared to males, a contrast to other major DCM genes such as TTN. These findings clarify sex-specific genetic architecture underlying DCM and highlight the importance of considering sex in genetic diagnosis, prognosis, and management. Advancing our understanding of sex-modulated penetrance will enhance precision medicine approaches in cardiovascular care and may lead to improved outcomes for high-risk female patients harboring DSP variants.
Future studies should aim to integrate environmental, hormonal, and reproductive factors alongside genetics to unravel the complex sex-specific pathophysiology of DCM.
References
- Tayal U, Ware JS, Lakdawala NK, Heymans S, Prasad SK. Understanding the genetics of adult-onset dilated cardiomyopathy: what a clinician needs to know. Eur Heart J. 2021;42(24):2384–2396.
- Mazzarotto F, Tayal U, Buchan RJ, et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation. 2020;141(5):387–398.
- Jordan E, Peterson L, Ai T, et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation. 2021;144(1):7–19.
- Pinto YM, Elliott PM, Arbustini E, et al. Proposal for a revised definition of dilated cardiomyopathy. Eur Heart J. 2016.

