Tóm tắt
– Các biến thể sense hiếm gặp trong CPD (carboxypeptidase D) liên quan đến mất thính lực bẩm sinh, chủ yếu là cảm giác thần kinh.
– Các biến thể gây bệnh làm suy giảm hoạt động xúc tác của CPD, giảm tín hiệu arginine/NO/cGMP, và thúc đẩy stress nội chất, tổn thương oxy hóa và apoptosis trong tế bào thính giác.
– Trong các mô hình động vật và tế bào, chức năng thính giác bị suy giảm do thiếu CPD được cải thiện một phần bằng cách bổ sung arginine hoặc tăng cường cGMP bằng thuốc (sildenafil), gợi ý một con đường điều trị có thể thực hiện được.
Nền tảng và bối cảnh lâm sàng
Mất thính lực bẩm sinh (HL) về mặt lâm sàng và di truyền là đa dạng, ảnh hưởng đến khoảng 1–3 trên 1.000 trẻ sơ sinh và mang lại hậu quả suốt đời đối với giao tiếp, phát triển và chất lượng cuộc sống. Nguyên nhân di truyền chiếm một tỷ lệ lớn của mất thính lực cảm giác thần kinh bẩm sinh; các biến thể gây bệnh trong nhiều gen (GJB2, SLC26A4, MYO7A, và các gen khác) thiết lập một phổ rộng các allel và cơ chế. Mặc dù chẩn đoán di truyền đã cải thiện, nhưng các liệu pháp y tế điều chỉnh bệnh cho HL di truyền vẫn còn ít, và hầu hết việc quản lý vẫn dựa vào trợ thính (máy trợ thính) hoặc phẫu thuật (implant ốc tai) và phục hồi chức năng. Các chiến lược dựa trên gen mới hoặc hướng dẫn con đường nhỏ phân tử là một nhu cầu chưa được đáp ứng quan trọng trong ngành tai mũi họng.
Thiết kế nghiên cứu và phương pháp (tóm tắt)
Nghiên cứu chính (Ramzan et al., J Clin Invest 2025) sử dụng một cách tiếp cận tích hợp giữa di truyền học con người và sinh học chức năng để liên kết mất chức năng CPD với HL bẩm sinh. Các yếu tố chính bao gồm:
- Xác định các biến thể sense CPD phân loại trong ba gia đình không liên quan với điếc bẩm sinh.
- Phân tích một nhóm lớn hơn (Dự án 100.000 Bộ gen) để kiểm tra xem có sự phong phú của các biến thể CPD hiếm gặp, thay đổi protein trong số những người mắc HL hay không.
- Nghiên cứu vị trí của Cpd trong ốc tai chuột để xác định biểu hiện của Cpd trong biểu mô cảm giác và các yếu tố thần kinh.
- Các thử nghiệm hóa sinh về hoạt động enzym của CPD và các sản phẩm chuyển hóa sau (arginine, nitric oxide (NO), cGMP) trong các sợi nguyên bào từ bệnh nhân.
- Các thử nghiệm tế bào và mô (biomarker stress nội chất, stress oxy hóa, apoptosis) trong các sợi nguyên bào và các nền văn hóa ốc tai organotypic sau khi làm im lặng Cpd.
- Các mô hình Drosophila để kiểm tra tác động lên cơ quan Johnston (cơ quan thính giác của ruồi), các kiểu hình hành vi/locomotor và các thí nghiệm cứu chữa dược lý với arginine hoặc sildenafil.
Kết quả chính
Di truyền và bằng chứng từ con người
Các nhà nghiên cứu đã phát hiện ra ba biến thể sense khác nhau ánh xạ đến vùng CP-domain2 hoạt động xúc tác của CPD trong năm cá nhân từ ba gia đình không liên quan với điếc bẩm sinh, chủ yếu đối xứng cảm giác thần kinh. Các biến thể này đồng phân loại với bệnh trong các gia đình và rất hiếm trong các cơ sở dữ liệu quần thể. Phân tích rộng hơn của bộ dữ liệu Dự án 100.000 Bộ gen đã tiết lộ sự phong phú thống kê của các biến thể CPD hiếm gặp, thay đổi protein trong số những người được ghi chú là mắc HL, hỗ trợ CPD là một gen có liên quan đến HL.
Vị trí và vai trò sinh lý
Trong ốc tai chuột, Cpd được biểu hiện ở cả các tế bào biểu mô cảm giác (bao gồm các khu vực tương ứng với tế bào lông và tế bào hỗ trợ) và các neuron trong đám rối xoắn. Mô hình biểu hiện này phù hợp với vai trò trong cả quá trình chuyển đổi cảm giác và cân bằng synaptical/neuronal trong đường dẫn thính giác.
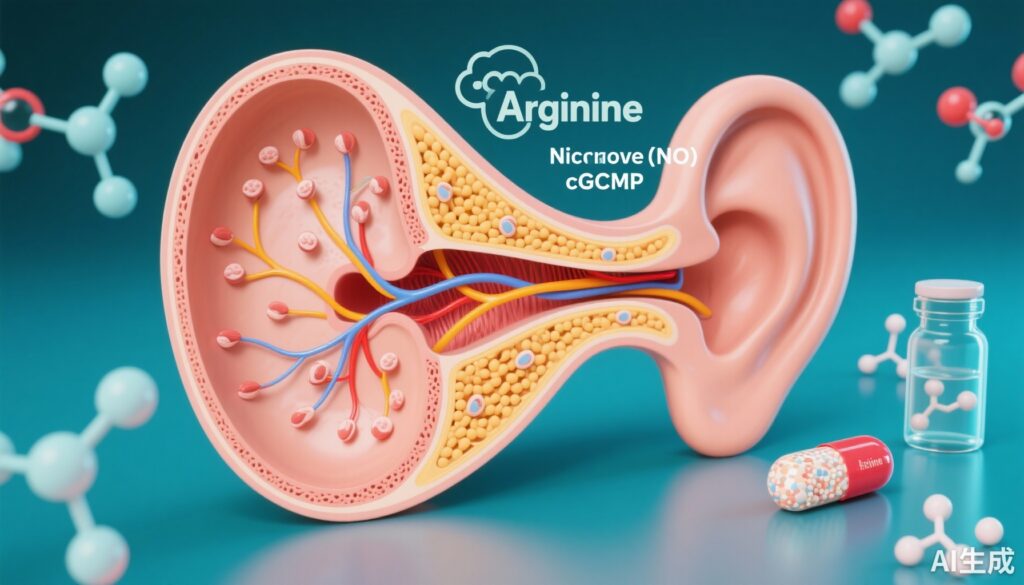
Cơ chế hóa sinh: trục arginine–NO–cGMP
CPD là một carboxypeptidase tham gia vào quá trình xử lý peptit; các tác giả đã chứng minh rằng các biến thể CPD được xác định làm giảm đáng kể hoạt động enzym. Trong các sợi nguyên bào từ bệnh nhân, nồng độ arginine, NO và cGMP thấp hơn so với đối chứng. Vì NO tổng hợp từ arginine kích hoạt guanylate cyclase hòa tan để tăng nồng độ cGMP trong tế bào, việc giảm tín hiệu NO/cGMP cung cấp một liên kết cơ chế hợp lý giữa việc suy giảm hoạt động CPD và sự rối loạn chức năng tế bào trong các mô thính giác.
Hậu quả tế bào: stress nội chất, stress oxy hóa, apoptosis
Các sợi nguyên bào từ bệnh nhân thể hiện các biomarker của stress nội chất (ER), stress oxy hóa tăng và tăng chết tế bào. Trong các nền văn hóa ốc tai organotypic của chuột, việc làm im lặng Cpd gây ra tăng apoptosis trong biểu mô cảm giác, chỉ ra các con đường chết tế bào tương tự trong mô ốc tai. Tổng thể, các dữ liệu này đề xuất một con đường từ việc suy giảm quá trình xử lý peptit/arginine đến việc gián đoạn tín hiệu NO, mất cân bằng redox và apoptosis do stress nội chất của các tế bào thính giác.
Mô hình in vivo và cứu chữa dược lý
Các mô hình Drosophila thiếu CPD chức năng cho thấy các khuyết tật cấu trúc trong cơ quan Johnston, suy giảm chuyển dẫn thính giác, và các kiểu hình hành vi/locomotor bị ảnh hưởng. Quan trọng, hai loại can thiệp đã cứu chữa một phần các kiểu hình trong ruồi và trong một số thử nghiệm ex vivo: bổ sung arginine (để phục hồi chất nền cho tổng hợp NO) và sử dụng sildenafil (một chất ức chế phosphodiesterase loại 5 tăng cGMP bằng cách giảm sự phân hủy của nó). Các can thiệp này không hoàn toàn bình thường hóa tất cả các chỉ số, nhưng sự cứu chữa một phần hỗ trợ giả thuyết rằng việc tăng cường tín hiệu NO–cGMP có thể cải thiện chức năng thính giác liên quan đến sự thiếu hụt CPD.
Giải thích và ý nghĩa dịch chuyển
Nghiên cứu này xác định CPD là một nguyên nhân di truyền mới của mất thính lực cảm giác thần kinh bẩm sinh và làm rõ một cơ chế có thể can thiệp — suy giảm tổng hợp NO phụ thuộc vào arginine với sự thất bại của tín hiệu cGMP, dẫn đến stress tế bào và apoptosis trong các mô thính giác. Từ góc độ dịch chuyển, hệ quả có thể thực hiện ngay nhất là các can thiệp sẵn có lâm sàng nâng cao tín hiệu NO/cGMP (bổ sung dinh dưỡng arginine và các chất tăng cường cGMP như sildenafil) có thể được sử dụng lại như các liệu pháp tiềm năng để thử nghiệm trong các giai đoạn thử nghiệm sớm. Sự cứu chữa một phần được quan sát trong các mô hình không xương sống và ex vivo cung cấp bằng chứng quan trọng về khái niệm nhưng chưa đủ để chứng minh hiệu quả tiền lâm sàng đầy đủ trên hệ thống thính giác của động vật có vú.
Bình luận chuyên gia, lưu ý và hạn chế
Lợi thế của nghiên cứu bao gồm sự kết hợp giữa di truyền học con người, phân tích phong phú quy mô quần thể, hóa sinh cơ chế và xác nhận chức năng xuyên loài. Việc xác định một con đường chuyển hóa-tín hiệu cụ thể sau CPD tăng cường tính hợp lý sinh học và mở ra một hướng điều trị hợp lý thay vì để gen chỉ là một sự tò mò chẩn đoán.
Các hạn chế và điểm cần thận trọng:
- Số lượng bệnh nhân nhỏ và phổ hiện tượng chưa được xác định. Tỷ lệ thâm nhập, tiến triển và mức độ nghiêm trọng khác nhau của các allele CPD khác nhau yêu cầu các nghiên cứu nhóm lớn hơn và dữ liệu lịch sử tự nhiên dự kiến.
- Các sợi nguyên bào dễ tiếp cận nhưng không phải là tế bào lông ốc tai hoặc neuron đám rối xoắn; việc suy diễn từ các dòng tế bào ngoại biên đến sinh lý ốc tai có giới hạn cố hữu. Trong khi các nền văn hóa ốc tai organotypic củng cố mối liên kết, dữ liệu cứu chữa in vivo đầy đủ trên động vật có vú (mô hình gen chuột) sẽ là bước tiếp theo quan trọng.
- Bổ sung arginine và chất ức chế PDE5 có tác động toàn thân. An toàn, liều lượng tối ưu, thời điểm (sơ sinh so với thời thơ ấu sớm so với sau này), và các hậu quả ngoài mục tiêu tiềm ẩn phải được xác định trước khi sử dụng lâm sàng, đặc biệt là ở trẻ sơ sinh và trẻ nhỏ.
- Sự cứu chữa một phần trong các mô hình gợi ý rằng cửa sổ điều trị có thể hẹp (việc ngăn ngừa chết tế bào có thể yêu cầu can thiệp sớm) và có thể cần các chiến lược kết hợp hoặc hướng gen để phục hồi hoàn toàn.
Ý nghĩa lâm sàng và nghiên cứu — một con đường đề xuất
Các ưu tiên ngay lập tức bao gồm:
- Sàng lọc di truyền rộng rãi hơn các nhóm HL bẩm sinh để xác định tần suất và phổ allele của các biến thể CPD.
- Phát triển các mô hình gõ ra hoặc gõ vào Cpd ở chuột để đánh giá biểu hiện dài hạn và thử nghiệm điều trị tiền lâm sàng, bao gồm dược động học và an toàn của arginine và sildenafil ở động vật có vú non.
- Xác định và xác nhận các biomarker (ví dụ, mức độ NO/cGMP hệ thống hoặc cục bộ, các dấu hiệu stress nội chất) để lựa chọn bệnh nhân và theo dõi hiệu quả dược động học.
- Thiết kế một thử nghiệm lâm sàng giai đoạn sớm được lên kế hoạch cẩn thận (giai đoạn 1/2) ở bệnh nhân có CPD thiếu hụt được xác nhận di truyền: các mục tiêu ngắn hạn có thể bao gồm an toàn, khả năng chịu đựng và điều chỉnh biomarker; các điểm cuối hiệu quả nên kết hợp các biện pháp audiometric khách quan (phản ứng não gốc thính giác, phát âm tai) với các kết quả phát triển/chức năng.
Kết luận
Ramzan et al. (J Clin Invest 2025) thêm CPD vào danh sách các gen gây điếc ở người và, quan trọng hơn, tiết lộ một con đường cơ chế — arginine–NO–cGMP — có thể can thiệp bằng các liệu pháp hiện tại. Điều này thay đổi mô hình cho ít nhất một phần của mất thính lực di truyền từ chỉ chẩn đoán sang có thể điều trị, và minh họa một quỹ đạo dịch chuyển nơi chẩn đoán phân tử có thể trực tiếp hướng dẫn các liệu pháp tái sử dụng. Tuy nhiên, nhiều bước vẫn còn phải thực hiện trước khi áp dụng lâm sàng: các nghiên cứu di truyền lớn hơn, các mô hình tiền lâm sàng ở động vật có vú, hồ sơ an toàn, và các thử nghiệm lâm sàng được thiết kế cẩn thận để xác định hiệu quả, thời điểm và kết quả dài hạn.
Quỹ tài trợ và thử nghiệm lâm sàng
Nguồn tài trợ và công nhận chi tiết được báo cáo trong bài báo gốc (Ramzan et al., J Clin Invest 2025). Tại thời điểm xuất bản, không có thử nghiệm lâm sàng đã đăng ký về arginine hoặc chất ức chế PDE5 cụ thể cho mất thính lực liên quan đến CPD; việc đăng ký thử nghiệm sẽ được yêu cầu trước khi thử nghiệm trên người.
Tài liệu tham khảo
1. Ramzan M, Ortiz-Vega N, Zafeer MF, Lobato AG, Atik T, Abad C, Vadgama N, Duman D, Bozan N, Avcı Durmuşalioǧlu E, et al. Carboxypeptidase D deficiency causes hearing loss amenable to treatment. J Clin Invest. 2025 Sep 30:e192090. doi: 10.1172/JCI192090. Epub ahead of print. PMID: 41026541.
2. Smith RJH, Bale JF Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–890. doi:10.1016/S0140-6736(05)70853-5.
3. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature Genetics. 1997;15(4): 444–447. doi:10.1038/ng0497-444.
Ghi chú của tác giả
Bài báo này tóm tắt và diễn giải các kết quả được báo cáo bởi Ramzan et al. và đặt chúng trong bối cảnh lâm sàng và dịch chuyển. Các bác sĩ không nên thay đổi cách quản lý bệnh nhân dựa chỉ trên các báo cáo tiền lâm sàng; tư vấn di truyền và chăm sóc đa ngành vẫn là cần thiết cho các gia đình bị ảnh hưởng bởi mất thính lực bẩm sinh.