亮点
– 罕见的CPD(羧肽酶D)错义变异与先天性、主要是感音神经性听力损失相关。
– 致病性变异会损害CPD催化活性,降低精氨酸/NO/cGMP信号传导,并促进听觉细胞中的内质网应激、氧化损伤和细胞凋亡。
– 在动物和细胞模型中,通过补充精氨酸或药物增强cGMP(西地那非),部分挽救了由CPD缺乏引起的听觉功能障碍,表明了一条可干预的治疗路径。
背景和临床背景
先天性听力损失(HL)在临床上和遗传上具有异质性,影响约每1,000名新生儿中的1-3人,并对沟通、发展和生活质量产生终身影响。遗传因素是先天性感音神经性聋的主要原因之一;许多基因(如GJB2、SLC26A4、MYO7A等)的致病性变异确立了广泛的等位基因和机制谱系。尽管遗传诊断有所改进,但针对遗传性HL的疾病修饰医疗疗法仍然很少,大多数管理仍依赖助听器或人工耳蜗植入等外科手术和康复治疗。新的基于基因或通路导向的小分子策略是耳科学的重要未满足需求。
研究设计和方法(概要)
主要研究(Ramzan等,J Clin Invest 2025)采用整合人类遗传学和功能生物学的方法,将CPD功能丧失与先天性HL联系起来。关键要素包括:
- 在三个无关家庭的先天性聋患者中鉴定出分离的错义CPD变异。
- 分析更大队列(100,000基因组项目)以测试HL个体中罕见的蛋白质改变CPD变异是否富集。
- 在小鼠耳蜗中进行定位研究,绘制Cpd在感觉上皮和神经元元素中的表达。
- 对患者来源的成纤维细胞进行CPD酶活性和下游代谢物(精氨酸、一氧化氮(NO)、cGMP)的生化检测。
- 在Cpd沉默后的成纤维细胞和耳蜗类器官培养物中进行细胞和组织检测(内质网应激标志物、氧化应激、细胞凋亡)。
- 使用果蝇模型探讨对约翰斯顿器官(果蝇听觉器官)、行为/运动表型的影响以及药理学挽救实验(补充精氨酸或西地那非)。
关键发现
遗传学和人类证据
研究人员在三个无关家庭的五个患有先天性、大部分对称性感音神经性聋的个体中发现了三种不同的映射到CPD催化活性CP-domain2区域的错义变异。这些变异在家族中与疾病共分离,并且在人群数据库中罕见。对100,000基因组项目数据集的广泛分析显示,HL参与者中罕见的蛋白质改变CPD变异显著富集,支持CPD作为真正的HL相关基因。
定位和生理作用
在小鼠耳蜗中,Cpd定位于感觉上皮细胞(包括与毛细胞和支持细胞相对应的区域)和螺旋神经节中的神经元。这种表达模式与听觉通路中感觉传导和突触/神经元稳态的作用一致。
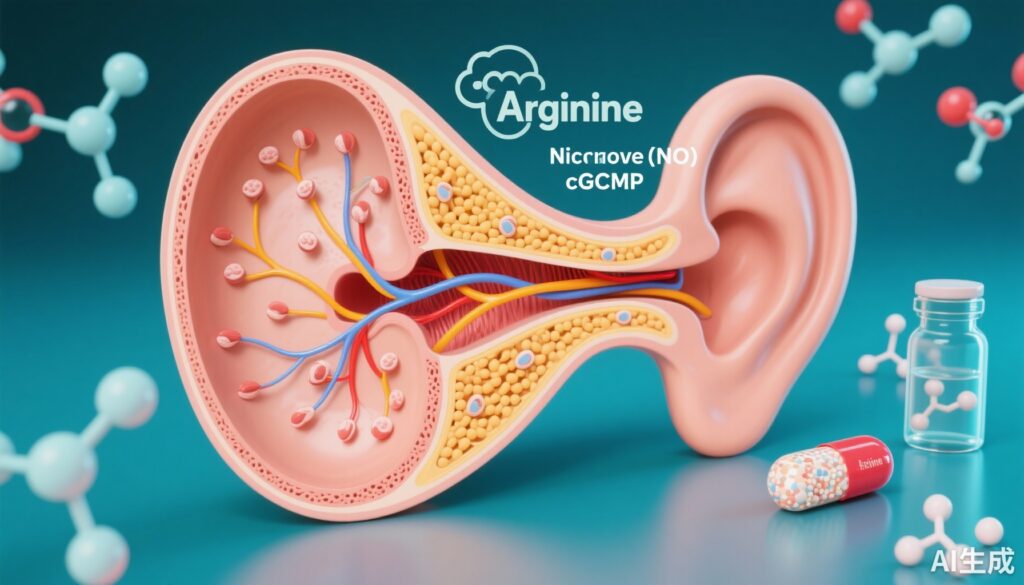
生化机制:精氨酸–NO–cGMP轴
CPD是一种参与肽加工的羧肽酶;作者显示,鉴定出的CPD变异显著降低了酶活性。来自受影响个体的成纤维细胞中,精氨酸、NO和cGMP的水平低于对照组。由于NO由精氨酸合成并通过激活可溶性鸟苷酸环化酶提高细胞内cGMP,因此NO/cGMP信号传导的减少为CPD活性受损与听觉组织细胞功能障碍之间的机制联系提供了合理的解释。
细胞后果:内质网应激、氧化应激、细胞凋亡
患者成纤维细胞表现出内质网(ER)应激的生物标志物、升高的氧化应激和增加的细胞死亡。在小鼠耳蜗类器官培养物中,沉默Cpd后,感觉上皮中的细胞凋亡增加,这表明相同的细胞死亡途径也存在于耳蜗组织中。综合这些数据表明,从受损的肽/精氨酸处理到NO信号传导中断、氧化失衡和内质网应激介导的听觉细胞凋亡的路径。
体内建模和药理学挽救
缺乏功能性CPD的果蝇模型显示出约翰斯顿器官的结构缺陷、听觉传导受损和感觉驱动的行为改变。重要的是,两种类型的干预措施部分挽救了果蝇和某些离体检测中的表型:补充精氨酸(以恢复NO合成的底物)和给予西地那非(一种通过减少其分解来增加cGMP的磷酸二酯酶5抑制剂)。这些干预措施并未完全恢复正常的所有指标,但部分挽救支持了增强NO–cGMP信号传导可以缓解CPD缺乏相关的听觉功能障碍的假设。
解释和转化意义
这项工作确立了CPD作为先天性感音神经性听力损失的新遗传原因,并阐明了一条可干预的机制——受损的精氨酸依赖性NO合成及其下游cGMP信号传导失败,导致听觉组织中的细胞应激和细胞凋亡。从转化角度来看,最直接可行动的后果是,现有的、临床上可用的提高NO/cGMP信号传导的干预措施(营养性精氨酸补充和药物cGMP增强剂如西地那非)可以作为候选疗法,在早期阶段试验中进行测试。在无脊椎动物和离体模型中观察到的部分挽救提供了重要的概念验证,但在哺乳动物听觉系统中尚不足以证明全面的临床前疗效。
专家评论、注意事项和局限性
研究的优势包括结合了人类遗传学、大规模人群富集分析、机制生化和跨物种功能验证。识别出CPD下游的特定代谢-信号传导通路增强了生物学可信度,并开启了一条合理的治疗途径,而不仅仅是将其作为一个诊断上的好奇。
关键的局限性和注意事项:
- 患者数量较少,表型谱尚未明确。不同CPD等位基因的外显率、进展和严重程度的变化需要更大的队列研究和前瞻性自然史数据。
- 成纤维细胞易于获取,但不是耳蜗毛细胞或螺旋神经节神经元;从外周细胞系推断内耳生理学存在固有的限制。虽然耳蜗类器官培养物加强了这一联系,但完整的哺乳动物体内挽救数据(啮齿动物基因敲除或敲入模型)将是重要的下一步。
- 精氨酸补充和PDE5抑制剂具有全身效应。在临床使用之前,特别是在婴儿和幼儿中,必须确定安全性、最佳剂量、时机(新生儿期、早期儿童期或后期)以及潜在的脱靶后果。
- 模型中的部分挽救表明治疗窗口可能较窄(预防细胞死亡可能需要早期干预),并且为了完全恢复可能需要组合或基因靶向策略。
临床和研究意义——建议的前进方向
立即优先事项包括:
- 更广泛地对先天性HL队列进行遗传筛查,以确定CPD变异的患病率和等位基因谱。
- 开发啮齿动物Cpd基因敲除或敲入模型,用于纵向表型分析和临床前治疗测试,包括在未成熟哺乳动物中精氨酸和西地那非的药代动力学和安全性。
- 识别和验证生物标志物(如全身或局部NO/cGMP水平、内质网应激特征),以选择患者并监测药效学效应。
- 设计一个精心分阶段的早期临床试验(1/2期),在遗传确认的CPD缺乏患者中进行:短期目标可以包括安全性、耐受性和生物标志物调节;有效性终点应结合客观听力测量(听觉脑干反应、耳声发射)与发展/功能结果。
结论
Ramzan等(J Clin Invest 2025)将CPD添加到人类聋基因目录中,并且关键地揭示了一条可通过现有疗法干预的机制通路——精氨酸–NO–cGMP。这改变了至少一部分遗传性听力损失的范式,从纯诊断转向潜在可治疗,并展示了分子诊断可以直接指导再利用疗法的转化轨迹。然而,在临床应用之前,仍有多步骤需要完成:更大规模的遗传研究、哺乳动物临床前模型、安全性分析以及精心设计的临床试验以确定疗效、时机和长期结果。
资金和临床试验
资金来源和详细致谢已在原始文章(Ramzan等,J Clin Invest 2025)中报告。截至出版时,尚未知有注册的针对CPD相关听力损失的精氨酸或PDE5抑制剂的临床试验;在人体测试之前需要进行试验注册。
参考文献
1. Ramzan M, Ortiz-Vega N, Zafeer MF, Lobato AG, Atik T, Abad C, Vadgama N, Duman D, Bozan N, Avcı Durmuşalioǧlu E, et al. Carboxypeptidase D deficiency causes hearing loss amenable to treatment. J Clin Invest. 2025 Sep 30:e192090. doi: 10.1172/JCI192090. Epub ahead of print. PMID: 41026541.
2. Smith RJH, Bale JF Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–890. doi:10.1016/S0140-6736(05)70853-5.
3. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature Genetics. 1997;15(4): 444–447. doi:10.1038/ng0497-444.
作者注
本文总结并解读了Ramzan等报告的发现,并将其置于临床和转化背景下。临床医生不应仅根据临床前报告改变患者管理;对于受先天性听力损失影响的家庭,遗传咨询和多学科护理仍然是必不可少的。